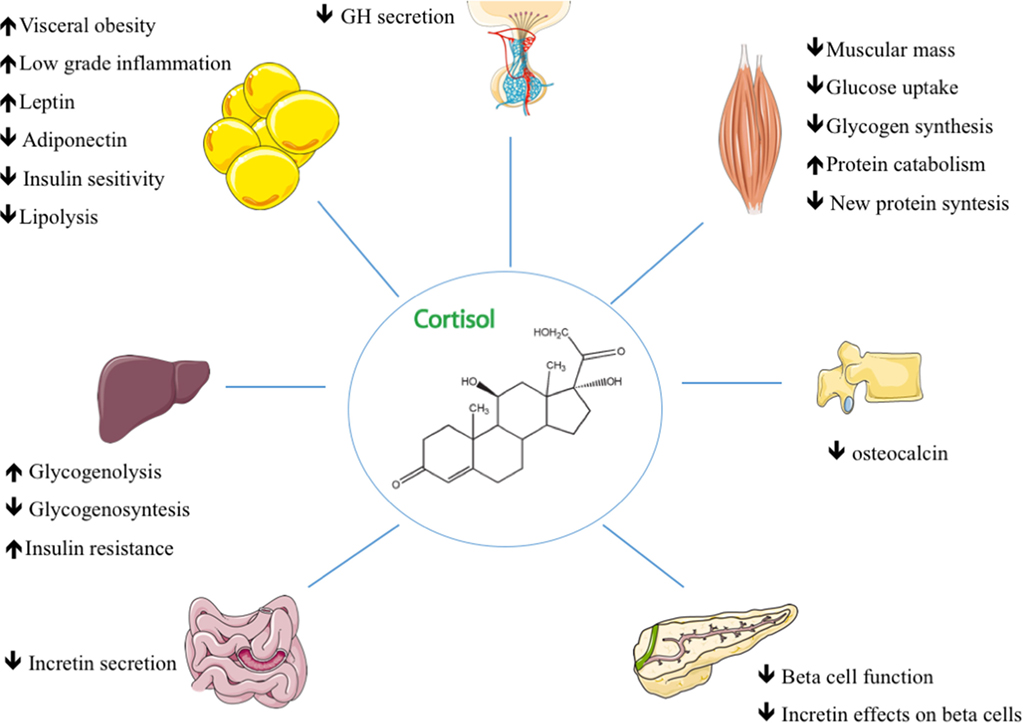
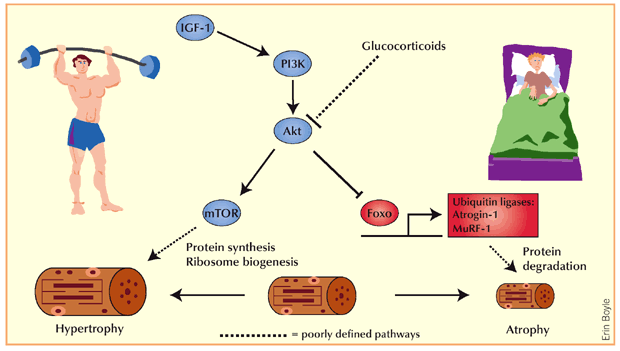
Introduzione alla Parte 3:
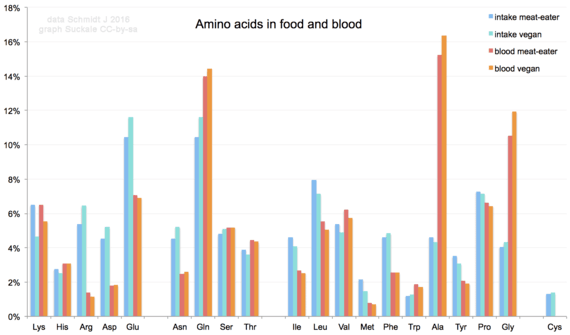
Nella 2° parte abbiamo analizzato le caratteristiche e funzioni biochimiche della Glutammina. In questa terza parte, invece, andremo ad analizzare due AA legati tra loro per via metabolica, la L-Citrullina e la L-Arginina.
Dalla L-Citrullina alla L-Arginina – Biologia e principali attività:

Il composto organico Citrullina è un α-amminoacido (formula H2NC(O)NH(CH 2)3CH(NH2)CO2H. ).[1] Sebbene sia stato nominato e descritto dai gastroenterologi fin dalla fine del XIX secolo, è stato isolato per la prima volta dall’anguria nel 1914 dai ricercatori giapponesi Yotaro Koga e Ryo Odake [2] [3] e ulteriormente codificato da Mitsunori Wada dell’Università Imperiale di Tokyo nel 1930.[4] La L-Citrullina è un composto aminoacidico non proteico (non viene utilizzato per formare proteine strutturali come gli enzimi) e, a differenza della L-Arginina, non è ampiamente presente in tutte le proteine. Si trova in concentrazioni particolarmente alte nell’anguria (da cui deriva il suo nome, dato che i cocomeri sono conosciuti come Citrullus vulgaris[1]), dove si trova in media a 2,1mg/g di peso umido (anche se i numeri assoluti variano)[2] e si è notato che il consumo di anguria aumenta in modo acuto sia l’Arginina plasmatica che la Citrullina (3.3 kg di anguria equivalgono a 10g di L-Arginina supplementare)[3][4] e di aumentare l’Arginina e l’Ornitina a digiuno del 12-22% in seguito al consumo di 780-1560g al giorno.[5]
Altre fonti alimentari di L-Citrullina sono i meloni, i meloni amari, le zucchine, le zucche e i cetrioli.[6]
La Citrullina viene sintetizzata nell’organismo attraverso una delle due vie: riciclata dall’Arginina (la conversione dell’arginina in ossido nitrico lascia la citrullina come sottoprodotto)[7][8] o prodotta dall’azoto (e da una parte del carbonio) contenuto nella L-glutammina,[9] dove l’enzima ornitina transcarbamilasi utilizza sia l’Ornitina che il carbamoilfosfato (che richiede la Clutammina) per produrre Citrullina negli enterociti.[10][11]
Sembra che la via dell’Arginina sia responsabile di circa il 10% della Citrullina circolante, mentre la via della Glutammina ne rappresenta il 90%;[6] la riduzione dei livelli plasmatici di Glutammina può ridurre la Citrullina plasmatica.[12]

Per quanto riguarda il ciclo dell’urea (uno dei meccanismi alla base del 10%), la L-Arginina viene convertita in L-Ornitina tramite l’enzima arginasi (cedendo urea come cofattore)[13][14] e da qui l’Ornitina (utilizzando il carbamoilfosfato come cofattore) viene sottoposta all’enzima Ornitina carbamoiltransferasi per produrre L-Citrullina. In questo senso, la via metabolica dall’Arginina alla Citrullina (attraverso l’Ornitina) provoca un aumento dell’urea e una concomitante diminuzione dell’ammoniaca, utilizzata dall’enzima carbamoilfosfato sintasi per creare carbamoilfosfato.[15] Se necessario, l’arginina può essere convertita direttamente in L-Citrullina attraverso un enzima arginina deiminasi per produrre, anziché richiedere, ammoniaca.[16]
Il ciclo si forma quando la citrullina si lega con l’L-aspartato (correlato all’acido D-aspartico come isomero) per formare l’arginosuccinato attraverso l’enzima arginosuccinato sintasi, quindi l’enzima arginosuccinato lisasi degrada l’arginosuccinato in arginina libera e fumarato; l’arginina rientra quindi nel ciclo dell’urea. [Il fumarato può semplicemente entrare nel ciclo TCA (Krebs) come intermedio energetico,[17] e la citrullina regola negativamente l’enzima arginasi.[18]
Anche la conversione della citrullina in L-arginosuccinato e la successiva conversione in L-arginina è coinvolta nel ciclo dell’ossido nitrico piuttosto che nel ciclo dell’urea, con l’unica differenza che l’arginina si converte direttamente in citrullina (cedendo una molecola di ossido nitrico) piuttosto che essere convertita indirettamente tramite l’ornitina.[18][19]
Come accennato, l’Arginina entra prima in contrata con il metabolismo intestinale e splancnico, in cui una certa quantità di essa viene consumata dagli enterociti o interconvertita in L-citrullina o L-ornitina. Oltre all’elevato utilizzo dell’arginina da parte del fegato, anche l’assorbimento intestinale dell’arginina è scarso in condizioni normali e aumenta in varie patologie.[20] Sembra che una quantità minima di L-arginina arrivi ai tessuti sistemici rispetto agli altri aminoacidi del ciclo dell’urea, dato che la L-ornitina supplementare raggiunge una concentrazione sierica doppia rispetto alla L-arginina e la L-citrullina 9,3 volte superiore. Ciò sembra direttamente correlato al grado di metabolismo epatico e intestinale.[21][22][23]

L’Arginina alimentare rappresenta il 40-60% dell’arginina sierica, come evidenziato da un calo equivalente durante i periodi di assenza di arginina. Il tasso di conversione della L-citrullina in L-arginina non sembra influenzato dall’assunzione con la dieta.[24]
La citrullina di per sé è più che altro un sottoprodotto del metabolismo dell’arginina (ciclo dell’ossido nitrico) e dell’ornitina (ciclo dell’urea) e viene semplicemente riconvertita in arginina tramite l’arginosuccinato. Detto questo, l’integrazione di citrullina influisce positivamente anche sulle concentrazioni di arginina e ornitina, quindi anche la loro bioattività è rilevante.
L’arginina può essere convertita in L-citrullina attraverso gli enzimi dell’ossido nitrico sintasi (NOS), di cui esistono forme endoteliali (eNOS) e neuronali specifiche (nNOS), nonché una forma inducibile (iNOS) che risponde ai segnali infiammatori. La conversione dell’arginina attraverso gli enzimi NOS produce ossido nitrico come sottoprodotto più importante, e la Citrullina è vista come un sottoprodotto.[25] La Citrullina può poi riconvertirsi in L-arginina attraverso l’arginosuccinato, ma la L-ornitina non è coinvolta nella via dell’ossido nitrico.
La L-Citrullina viene assorbita nell’intestino in misura molto maggiore rispetto alla sua controparte L-Arginina e determina un livello plasmatico più elevato di L-Arginina attraverso il ciclo Arginina/Ornitina/Citrullina.[26] Viene assorbita attraverso numerosi trasportatori sodio-dipendenti.[27]
È stato osservato che l’integrazione orale di citrullina nell’uomo a 0,18 g/kg raddoppia l’arginina plasmatica[28], cosa che è stata replicata altrove[29], insieme a un aumento equivalente delle concentrazioni di ornitina[29], ma questi raddoppi di arginina e ornitina sono associati a un aumento di 6-11 volte della citrullina plasmatica[28][29].
Una singola dose di 6 g di citrullina malato (0,08 g/kg) in atleti prima dell’esercizio fisico ha fatto registrare aumenti della citrullina plasmatica (aumento del 173%), dell’ornitina (aumento del 152%) e dell’arginina (aumento del 123%) quando misurata dopo l’esercizio fisico, valori che si sono normalizzati con 3 ore di riposo.[30] Questa stessa dose è stata notata altrove per aumentare la citrullina plasmatica e l’arginina in misura simile.[31]
È interessante notare che gli studi sopra citati che hanno utilizzato 0,18 g/kg di citrullina hanno rilevato un aumento di 6-11 volte della citrullina a fronte di un mero raddoppio dell’arginina e dell’ornitina[28][29], mentre lo studio successivo che ha utilizzato 6 g (calcolati come 0,08 g/kg) ha registrato un aumento molto minore della citrullina, ma ha comunque più che raddoppiato sia l’arginina che l’ornitina. [30] Ciò è stato osservato anche in uno studio dose-risposta che ha utilizzato da 2 a 15 g di citrullina, in cui la citrullina nel plasma ha seguito una dipendenza lineare dalla dose, mentre sia l’arginina che l’ornitina hanno avuto una dipendenza minore dalla dose.[29] Gli autori hanno ipotizzato che, dato che l’aumento dell’arginina è stato inferiore a quello previsto e che la citrullina sierica è il principale predittore della sintesi dell’arginina[19], ciò indichi il raggiungimento di una fase di limitazione della velocità nei reni.
È stato osservato che la citrullina non influenza i livelli sierici degli aminoacidi a catena ramificata a riposo,[21] ma può accelerare la deplezione dei BCAA indotta dall’esercizio fisico prolungato (aumentandone l’utilizzo come carburante).[20]
Con l’integrazione di citrullina è stata notata una riduzione della glutammina (13% dopo 0,18 g/kg di citrullina per 7 giorni)[21], anche se un altro studio ha rilevato che l’uso acuto di 6 g di citrullina (0,08 g/kg) non ha alterato le concentrazioni di glutammina.[20]
Gli altri aminoacidi testati (acido glutammico, acido aspartico, asparagina, alanina, lisina, triptofano, fenilalanina, L-tirosina, istidina) sono per lo più inalterati.[20]
Circa l’83% della citrullina ingerita per via orale sembra essere assorbita dai reni[26][27][28] dove viene convertita in L-arginina nei tubuli prossimali (attraverso gli enzimi arginosuccinato sintasi e arginosuccinato liasi[29]); Questa conversione della citrullina in arginina (sia da citrullina supplementare che da quella prodotta come sottoprodotto della creazione di ossido nitrico da parte dell’arginina) rappresenta il 5-15% dell’arginina circolante. [11][30]
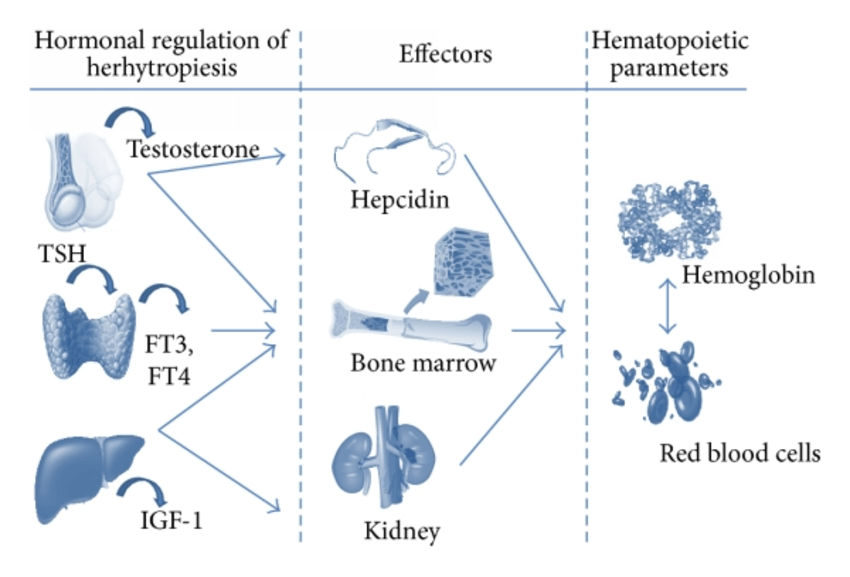
Il meccanismo principale con cui l’integrazione di arginina (e, per estensione, di L-citrullina) influisce sulla salute del sangue è quello di essere il substrato per gli enzimi dell’ossido nitrico sintasi (NOS) per la produzione di ossido nitrico, che poi segnala attraverso i recettori ciclici solubili della guanilina la produzione di cGMP. La produzione di ossido nitrico e la successiva produzione di cGMP intracellulare sono alla base di buona parte dei benefici dell’arginina.
Gli enzimi NOS si presentano in tre isoforme principali: [32][33] la NOS inducibile (iNOS), che viene creata in risposta a fattori di stress infiammatori, la NOS neuronale (nNOS), che è stata scoperta per la prima volta nei neuroni e si trova anche nelle terminazioni motorie dei muscoli scheletrici, e la NOS endoteliale (eNOS), che inizialmente si pensava si trovasse solo nell’endotelio, ma è piuttosto diffusa[34], compreso il tessuto cerebrale.[35][36]

Gli enzimi NOS lavorano in dimeri uniti testa a testa e i meccanismi catalitici dipendono da questa dimerizzazione, oltre che dall’eme, dalla tetraidrobiopterina, dalla calmodulina, dal NADPH (come donatore di elettroni) e da FMN e FAD.[37][38][39] Di conseguenza, gli enzimi NOS (tutte e tre le isoforme) sono flavoproteine che richiedono NADPH. [40][41][42] La loro struttura e funzione è complessa (esaminata qui[43]), ma esiste un sito di legame di base per l’arginina e gli elettroni donati dal NADPH fanno sì che l’arginina si converta in citrullina, rilasciando come sottoprodotto l’ossido nitrico; l’iNOS utilizza esclusivamente e l’eNOS può anche utilizzare un intermedio radicale libero chiamato Nω-idrossi-L-arginina (L-NOHA), che si degrada in citrullina e ossido nitrico in presenza di H2O2.[32][44]
L’aumento dell’ossido nitrico (solitamente misurato attraverso le concentrazioni plasmatiche di nitrato/nitrito, citrullina o cGMP urinario) sembra essere aumentato con la L-arginina in persone affette da ipertensione essenziale,[45] arterotrombosi,[46] e diabete di tipo II. [47] Gli studi condotti su atleti altrimenti sani che assumono L-arginina sono piuttosto contrastanti; ci sono casi in cui i biomarcatori del metabolismo dell’ossido nitrico sono aumentati[48] mentre altri studi non notano alcuna modifica.[49][50][51] Non sorprende che i benefici associati all’ossido nitrico non si verifichino quando i biomarcatori dell’ossido nitrico non sono aumentati.
L’inaffidabilità dell’aumento dell’ossido nitrico da parte dell’arginina può essere dovuta al fatto che le concentrazioni fisiologiche di arginina (40-100µM nello spazio extracellulare[52] e forse fino a 800µM a livello intracellulare[53]) sono sufficienti a saturare intrinsecamente l’ossido nitrico sintasi endoteliale (eNOS) (di solito si dichiara una Km di 3µM[54][55], ma a volte viene misurata fino a 29μM[56). Ciò implica che l’enzima è già al massimo dell’efficacia e che un’ulteriore integrazione non aumenta il tasso di conversione (a causa di un arretrato di arginina nel siero); l’osservazione che l’arginina aumenta ancora l’ossido nitrico a volte (anche se in modo inaffidabile) è indicata come il paradosso della L-arginina.[57][58]
Questa teoria è in linea con le osservazioni secondo cui a volte il metabolismo dell’ossido nitrico non viene influenzato nonostante aumenti fino al 300% dell’arginina plasmatica.[59]
Uno studio ha osservato un aumento transitorio della produzione di ossido nitrico che sembra essere più simile a quello di un agonista che di un substrato[60] e successivamente è stato scoperto che l’arginina ha la capacità di attivare i recettori α2-adrenergici,[61] che possono stimolare direttamente l’ossido nitrico senza richiedere la conversione in citrullina attraverso la NOS. Tuttavia, l’arginina è risultata piuttosto debole (superata dall’agmatina)[61] ma questo meccanismo non è ancora stato escluso.
Inoltre, l’arginina extracellulare sembra essere un fattore determinante per il rilascio di ossido nitrico[56] (il trasportatore CAT1 che trasporta l’arginina è altamente associato alla eNOS[62] e l’inibizione dell’afflusso extracellulare impedisce l’attivazione della eNOS[63]), mentre la concentrazione intracellulare di arginina non sembra essere associata. [58] Poiché il trasporto è necessario, ma l’arginina intracellulare non è di per sé necessaria, si ritiene che la colocalizzazione di CAT1 con eNOS[62][64] possa svolgere un ruolo nella stimolazione dell’attività di eNOS.
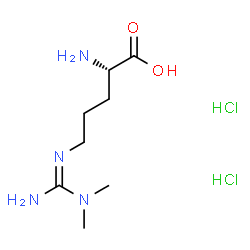
L’ADMA è un metabolita metilato dell’arginina e sembra agire in opposizione all’arginina inibendo le azioni dell’enzima NOS e la conseguente produzione di ossido nitrico. Livelli eccessivi di ADMA possono essere causati da fattori di stress ossidativo che diminuiscono l’attività dell’enzima che lo degrada, mentre la riduzione dell’ADMA provoca una vasodilatazione dovuta alla produzione di ossido nitrico.
Sebbene la maggior parte delle evidenze suggerisca che l’ADMA non aumenta con l’integrazione di L-arginina (questi studi notano che il rapporto arginina:ADMA è aumentato a causa dell’aumento dell’arginina plasmatica), ci sono prove limitate che suggeriscono un aumento che richiede ulteriori indagini.[38]
L’integrazione orale di arginina (anche la citrullina si applica in questo caso perché aumenta l’arginina plasmatica) è in grado di aumentare il flusso sanguigno nelle persone con flusso sanguigno ridotto e, sebbene abbia il potenziale di ridurre la pressione sanguigna, sembra un po’ inaffidabile e può verificarsi solo negli ipertesi.
Esistono prove contrastanti sugli effetti dell’integrazione di arginina sul flusso sanguigno in persone con resistenza periferica o cladicazione intermittente, con studi a breve termine che notano un beneficio e studi a più lungo termine che notano un’alterazione.[65][66]
L’integrazione di citrullina sembra ridurre la pressione sanguigna e migliorare il flusso sanguigno in situazioni in cui il flusso sanguigno è altrimenti ostacolato o la pressione sanguigna è più alta del normale, ma la citrullina non ha effetti di riduzione unidirezionali; può essere inefficace in persone normotese a riposo.[41]
In atleti allenati a cui sono stati somministrati 6 g di citrullina malato prima di un test ciclistico prolungato (137 km), l’aumento dell’ormone della crescita indotto dall’esercizio sembra essere aumentato; quando è stato misurato subito dopo l’esercizio, il gruppo con citrullina aveva concentrazioni di GH più elevate del 66,8%, che (dopo 3 ore di riposo) si sono attenuate al 28%.[20] Altrove, dosi di 2-15 g di citrullina non sono riuscite a influenzare l’ormone della crescita a riposo, se misurate nell’arco di 8 ore.[22]
Le concentrazioni di IGF-1 dopo 0,18 g/kg di citrullina per 7 giorni non sono state influenzate in modo significativo.
Durante l’esercizio fisico, sebbene uno studio che ha utilizzato 3 g di L-arginina (associata a 2.200 mg di L-ornitina e 12 mg di vitamina B12) per 3 settimane abbia rilevato un aumento del 35,7% della secrezione di ormone della crescita indotta dall’esercizio fisico (che si è normalizzata entro un’ora)[67], altri studi notano il contrario; è stato osservato che l’integrazione di arginina determina un minore picco di ormone della crescita durante l’esercizio fisico rispetto all’esercizio fisico da solo[68][69] e che, sebbene sembri influenzare maggiormente i giovani rispetto agli anziani[70], si dice che influisca su entrambi i gruppi di età. [L’entità della soppressione (supponendo che il 100% sia il valore di base) è stata notata intorno a un aumento del 300-500% visto con l’esercizio fisico, attenuato al 200%.[68]
È possibile che un aumento eccessivo dell’ormone della crescita stimoli un feedback autogeno, il che spiegherebbe come gli individui più anziani siano meno sensibili a questa soppressione, in quanto hanno intrinsecamente meno picchi di GH dovuti all’esercizio fisico rispetto ai giovani.[71] Infine, poiché i picchi dell’ormone della crescita si normalizzano comunque nel giro di poche ore[67][71], non si sa esattamente quanto sia preoccupante questa soppressione (dato che le concentrazioni di ormone della crescita nelle 24 ore sono più rilevanti).
A riposo, l’integrazione di 5-9 g di L-arginina è in grado di provocare un aumento delle concentrazioni di picco dell’ormone della crescita (aumento del 34,4-120%), mentre 13 g sono risultati inefficaci a causa della sofferenza intestinale che impedisce l’assorbimento della L-arginina.[36]
Negli studi che misurano la secrezione di GH nelle 24 ore, non sono state riscontrate alterazioni significative con la somministrazione di 2 g due volte al giorno[72] o con dosi acute di 5 g.[73] Ciò è potenzialmente legato a un noto fenomeno di feedback autonomo dell’ormone della crescita,[69][74][75] e un effetto modulatorio simile sull’ormone della crescita si riscontra anche durante la restrizione del sonno (in cui una riduzione del rilascio di ormone della crescita indotto dal sonno viene compensato durante le ore di luce).
È stato osservato che l’arginina ad alte dosi (250mg/kg di arginina aspartato al giorno, circa 17,5g di arginina) aumenta l’impulso di GH nel sonno a onde lente di circa il 60%, pur non avendo un’influenza sufficiente sulle concentrazioni di GH durante la veglia.[76] Non è chiaro come questo grande aumento influisca sulle concentrazioni di ormone della crescita nell’intera giornata.

L’integrazione di arginina nei ratti è in grado di aumentare il nitrato urinario post-esercizio, indicativo della produzione di ossido nitrico.[77] Aumenti nella produzione di ossido nitrico (nitrato urinario o sierico) sono stati confermati anche nell’uomo in seguito ad assunzione orale o infusione endovenosa.[78]
Non sempre si riscontra un aumento della produzione di ossido nitrico (anche nonostante l’aumento dell’arginina plasmatica), suggerendo che l’attività dell’enzima NOS potrebbe essere un fattore limitante.
Per quanto riguarda gli studi in acuto (assunzione di una singola dose di L-arginina prima dell’esercizio), 3 g di arginina (sotto forma di AAKG) non hanno apportato benefici all’allenamento con i pesi,[79] 6 g di L-arginina per 3 giorni non hanno modificato i risultati del cicloergometro in atleti di judo, mentre un protocollo simile in ciclisti allenati ha rilevato un miglioramento del tempo di esaurimento (25,8%).[80]
Alcuni studi hanno utilizzato una forma di arginina nota come GAKIC (Glycine L-Arginine α-Ketoisocaproic acid) e hanno rilevato un aumento della potenza media durante gli sprint di 10s su cicloergometro (con 11,2 g di GAKIC)[81] e un aumento del 10,5+/-0. Questi studi, tuttavia, sono confusi sia dall’inclusione della glicina sia da quella del metabolita della leucina, l’acido α-chetoisocaproico.
Per quanto riguarda gli studi più cronici, l’integrazione di L-arginina (come asparato) con 2,8 o 5,7 g di arginina al giorno per 4 settimane non è riuscita a modificare le prestazioni o altri biomarcatori[82]; anche uno studio precedente, condotto per 2 settimane con una metodologia simile, ha fallito.[83]
Nel complesso, quando si esaminano le revisioni o le meta-analisi sull’argomento L-arginina e prestazioni sportive, si nota che è promettente, ma manca un consenso sufficiente per raccomandarla come ergogenico.[84]
Si ritiene che la citrullina sia un agente pro-erettile in quanto è un precursore dell’arginina, e l’arginina è il substrato da cui viene prodotto l’ossido nitrico che può poi indurre il cGMP (attraverso la via NO/cGMP/VEGF);[65] un aumento del cGMP è anche l’effetto finale degli inibitori della PDE5 come il viagra o l’icariina dall’erba cornuta.[66]
Negli uomini con disfunzione erettile, valutata come debolezza dell’erezione (valutata con il punteggio di durezza yerettile[67]), la somministrazione di 1.500 mg di citrullina al giorno (due dosi da 750 mg) per un mese è stata in grado di apportare benefici alla metà dei 24 pazienti valutati (valutati come “molto soddisfatti” del trattamento), mentre il miglioramento del placebo è stato solo dell’8,3%.[68]
La citrullina sembra interagire con il metabolismo dei BCAA nell’organismo, anche se gli studi sull’uomo sembrano avere risultati diversi a seconda del contesto dello studio.
La citrullina può mediare positivamente la segnalazione della leucina attraverso mTOR, il che suggerisce teoricamente una sinergia. L’applicazione di questa combinazione ai sollevatori di pesi non è ancora stata studiata, quindi il sinergismo è attualmente solo un’ipotesi piuttosto che un fatto dimostrato.
Il nitrato è un piccolo donatore di ossido nitrico che costituisce il principale bioattivo del succo di barbabietola.
Il nitrito sierico (forma ridotta del nitrato) sembra aumentare durante l’esercizio fisico in seguito al consumo di 6 g di citrullina malato, che si ritiene sia un indicatore dell’aumento della produzione di ossido nitrico.[20]
I farmaci a base di statine possono aumentare l’espressione dell’enzima che media la conversione dell’arginina in ossido nitrico e per questo motivo è possibile che vi sia un sinergismo per tutto ciò che riguarda l’ossido nitrico. Questo non è ancora stato testato in un sistema vivente.
L-Citrullina come sostituto alla L-Arginina?
L’integrazione di L-citrullina è stata definita un’alternativa alla L-arginina, in quanto ne aggira lo scarso assorbimento e si converte in L-arginina nei reni. La L-citrullina tecnicamente segue aumenti dose-dipendenti della L-arginina sierica fino a 15 g, ma la dose orale più alta di citrullina assunta ha ritorni sempre minori (cioè per ogni 5 g in più di citrullina ingerita si aggiunge meno arginina al siero).[85]

È stato osservato che la citrullina orale a 0,18 g/kg raddoppia approssimativamente l’arginina plasmatica (aumento del 100%)[86][87] o è leggermente superiore (123%) con 0,08 g/kg.[88] Poiché le dosi più elevate di L-citrullina hanno una minore conversione in arginina[85], è improbabile che la L-citrullina supplementare possa essere utilizzata per superare l’arginina per l’aumento acuto dell’arginina sierica.
Gli studi che hanno confrontato direttamente la L-arginina con la L-citrullina hanno osservato che entrambe aumentano la Cmax a livelli comparabili a dosi orali simili (Cmax di 79+/-8μM per 3 g di citrullina e 84+/-9μM per l’arginina), ma la citrullina risulta in un’AUC complessiva maggiore (48,7% in più rispetto all’arginina). [Questo potrebbe essere dovuto al fatto che, anche fino all’ingestione di 15g di citrullina, non si verifica un aumento significativo dell’escrezione di citrullina.[85] L’assenza di un aumento dell’eliminazione di L-citrullina dal sangue nonostante l’integrazione consentirebbe di avere un pool di L-citrullina disponibile per la conversione su richiesta in L-arginina.
Citrullina Malato:
La Citrullina Malato (CM), una combinazione di L-citrullina e acido malico, è stata pubblicizzata come un aiuto ergogenico (che aumenta l’energia) per l’allenamento contro-resistenza e l’esercizio ad alta intensità.

Come abbiamo visto, la L-citrullina è un precursore dell’ossido nitrico (NO), un vasodilatatore che può migliorare l’apporto di sangue e ossigeno ai muscoli durante l’esercizio. Tuttavia, le prove finora disponibili suggeriscono che il miglioramento del flusso sanguigno non è il meccanismo attivo degli effetti ergogenici del CM. Il meccanismo potrebbe invece essere dovuto alla capacità della citrullina di favorire l’eliminazione dell’ammoniaca durante l’esercizio ad alta intensità, alla capacità del malato di aumentare la produzione di ATP, a un aumento dell’espressione genica o a una maggiore efficienza della navetta malato-aspartato.
La maggior parte delle ricerche condotte finora ha utilizzato una dose acuta di 8 grammi di CM un’ora prima dell’esercizio. Sebbene l’assunzione di CM un’ora prima dell’esercizio rimanga la migliore raccomandazione, alcuni dati suggeriscono che dosi maggiori, fino a 15 grammi, potrebbero essere più benefiche.
È stato dimostrato che l’ingestione di una serie di dosi di CM (2-15 grammi) non ha effetti negativi sui marker ematologici. Sebbene la sicurezza di un’integrazione di CM a lungo termine richieda ulteriori indagini, le ricerche condotte finora indicano che la CM è ben tollerata nella maggior parte degli individui.
Ricerche preliminari hanno suggerito che 8 grammi di CM ingeriti un’ora prima dell’esercizio fisico aumentano la resistenza muscolare (ripetizioni fino al cedimento) in uomini e donne. Tuttavia, ricerche più recenti hanno suggerito che il CM potrebbe non avere un beneficio sulle prestazioni nell’allenamento contro-resistenza, potenzialmente a causa di variazioni nei tempi e nei dosaggi.[89]
Arginina AKG:

La differenza principale tra la L-arginina e Arginina AKG è che la L-arginina è un aminoacido non essenziale che l’organismo è in grado di produrre, mentre l’arginina AKG è un sale della L-arginina e dell’acido α-chetoglutarato. Inoltre, la L-arginina regola il flusso sanguigno attraverso la produzione di ossido nitrico, mentre l’Arginina AKG dovrebbe potenzialmente aumentare il flusso sanguigno, l’energia e il recupero.
Nella nutrizione sportiva, l’AKG è stato utilizzato come integratore per migliorare la sintesi proteica muscolare e diminuire la disgregazione muscolare, ed è quindi utilizzato dagli atleti per migliorare la composizione corporea.[90][91] L’integrazione di AKG potrebbe anche migliorare le prestazioni atletiche. Uno studio ha rilevato che un integratore di arginina e alfa-chetoglutarato (AAKG) ha migliorato la forza nella panca, ma non la capacità aerobica. Sono necessarie ulteriori ricerche per sostenere le affermazioni sull’AKG come aiuto ergogenico.[92]
L’AKG viene utilizzato anche per il recupero da interventi chirurgici o traumi, perché è un precursore dell’aminoacido glutammina. Sebbene la glutammina sia un aminoacido non essenziale, viene talvolta definita “condizionatamente essenziale” perché la quantità di glutammina di cui l’organismo ha bisogno per il recupero dopo un trauma significativo può superare la quantità che l’organismo è in grado di produrre. In questo caso, un integratore di AKG può aiutare il processo di recupero.[93][94]
L’AKG è stato proposto come integratore per la longevità; alcune ricerche condotte su vermi tondi, ratti e topi suggeriscono che potrebbe aumentare la durata della vita e ritardare l’insorgenza di malattie legate all’età, anche se gli studi clinici dovranno confermare questi risultati.[95][96]
Nelle persone con malattie renali croniche, in particolare in quelle sottoposte a dialisi, la somministrazione di AKG in combinazione con il calcio ha migliorato i biomarcatori della funzione renale.[97][98]
In uno studio è stato rilevato che l’AKG aumenta l’espressione di involucrina, filaggrina e serina palmitoil transferasi. Queste molecole sono tutte importanti per la struttura dello strato esterno della pelle e per l’idratazione dello strato esterno della pelle, quindi l’uso di AKG per via topica potrebbe migliorare l’aspetto della pelle.[99][100]
Nella ricerca, i dosaggi utilizzati variano da 3,6 g a 6 g, con dosaggi più elevati nelle persone che hanno subito ustioni, ma non è ancora stata stabilita una dose giornaliera raccomandata.[101] Poiché gli effetti sono dose-dipendenti, trovare una raccomandazione di dosaggio accurata sarà una parte importante della ricerca in corso.[102]
Sicurezza e tossicità:
La citrullina sembra essere ben tollerata dai ratti in dosi fino a 3 g/kg di peso corporeo[58][46].
Negli esseri umani, 15 g di citrullina assunti acutamente non sembrano causare diarrea o disturbi intestinali[22], il che è notevolmente diverso rispetto all’ornitina e all’arginina che possono causare diarrea a dosaggi di 10 g se assunti in bolo[74][75] a causa del limitato assorbimento di questi aminoacidi che poi procedono verso il colon causando diarrea osmotica.[74]
Il limite di sicurezza osservato, ovvero la dose più alta in cui si può essere relativamente sicuri che non si verifichino effetti collaterali nel corso della vita, è stato suggerito in 20 g di arginina al giorno in forma di integratore (al di sopra dell’assunzione di cibo).[103] Dosi più elevate sono state testate e ben tollerate, ma non esistono prove che suggeriscano la loro sicurezza in tutte le popolazioni nel corso della vita.
La L-arginina ha un tasso di assorbimento gastrointestinale piuttosto scarso. Può inoltre agire come assorbente, rilasciando acqua ed elettroliti nel lume intestinale attraverso la stimolazione dell’ossido nitrico e inducendo disturbi gastrici e diarrea.[12] Questo fenomeno è noto come diarrea osmolitica e tende a verificarsi a dosi orali superiori a 10 g circa, se assunte in bolo.[36]
Si pensa che ciò avvenga attraverso la stimolazione della produzione di ossido nitrico, poiché la D-Arginina (incapace di produrre NO) non produce diarrea[104] e l’ossido nitrico stesso è noto per essere un meccanismo attraverso il quale molti lassativi osmolitici funzionano.[105]
Singoli boli di 5-9 g di L-arginina senza cibo non sembrano causare disturbi intestinali come dosi superiori a 10 g,[36] suggerendo che, almeno per uno stomaco vuoto, il dosaggio di 9 g è un limite superiore.
Conclusioni:
L’Integrazione di L-citrullina si è dimostrata più redditizia tra costi e benefici (vedi assorbimento intestinale) rispetto alla L-arginina. L’uso alternativo di Citrullina Malato può portare ad eventi gastrointestinali più frequenti rispetto alla semplice forma L-citrullina. Nonostante la ridotta biodisponibilità orale della L-arginina, questa può essere mixata con L-citrullina per un effetto additivo, anche se non rappresenta un vero e proprio vantaggio proprio di tale abbinamento.
L’assunzione di L-arginina e/o L-citrullina vede la sua miglior tempistica prima dell’allenamento al fine di aumentare il flusso sanguigno ai distretti allenati, per effetto della vasodilatazione NO indotta.
Ciò si traduce in:
- Aumento dell’apporto di nutrienti e ossigeno al tessuto muscolare abbinato ad un effetto di pulizia dalle molecole di scarto, come l’acido lattico;
- Esaltazione dilatatoria sul reticolo venoso sottocutaneo, che migliora la qualità estetica in definizione.
Bonus: l’abbinamento con estratto di barbabietola notoriamente ricco di nitrati.
Come effetto diretto dell’introduzione nell’organismo di estratto di barbabietola abbiamo un aumento della sintesi di ossido nitrico (NOs), dovuta, per l’appunto, ai nitrati (NO3-) contenuti in questo vegetale, convertiti rapidamente in nitriti (NO2-2) tramite enzimi che si trovano fin dal tratto orofaringeo, gastrointestinale e tracheo-bronchiale. Dato ciò, la sintesi di NO sfrutta un percorso non usuale come quello della L-Arginina, ma coadiuvante a questa e alla L-Citrullina.

Le concentrazioni di nitrati raggiungono il picco dopo circa un’ora dalla sua ingestione, per ritornare ai livelli basali dopo quasi 24h, mentre gli effetti della L-Arginina permangono per almeno 75-80 minuti, per poi iniziare a tornare ai livelli basali.
Continua…
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
- Banerjee, Aryamitra (2014-01-01), Gupta, Ramesh C. (ed.), “Chapter 15 – Gastrointestinal toxicity biomarkers”, Biomarkers in Toxicology, Boston: Academic Press, pp. 269–277, doi:10.1016/b978-0-12-404630-6.00015-4, ISBN 978-0-12-404630-6, S2CID 88798984, retrieved 2020-11-10
- Fragkos, Konstantinos C.; Forbes, Alastair (September 2011). “Was citrulline first a laxative substance? The truth about modern citrulline and its isolation” (PDF). Nihon Ishigaku Zasshi. [Journal of Japanese History of Medicine]. 57 (3):
- “Citrulline – Compound Summary”. PubChem Compound. USA: National Center for Biotechnology Information. 16 September 2004. Identification. Retrieved 1 May 2012.
- Fearon, William Robert (1939). “The Carbamido Diacetyl Reaction: A Test For Citrulline”. Biochemical Journal. 33 (6): 902–907. doi:10.1042/bj0330902. PMC 1264464. PMID 16746990.
- “Nos2 – Nitric Oxide Synthase”. Uniprot.org. Uniprot Consortium. Retrieved 10 February 2015.
- Cox M, Lehninger AL, Nelson DR (2000).
- Curis E, Nicolis I, Moinard C, Osowska S, Zerrouk N, Bénazeth S, Cynober LAlmost all about citrulline in mammalsAmino Acids.(2005 Nov)
- Metabolism of citrulline in man.()
- Tomlinson C, Rafii M, Ball RO, Pencharz PArginine synthesis from enteral glutamine in healthy adults in the fed stateAm J Physiol Endocrinol Metab.(2011 Aug)
- van de Poll MC, Ligthart-Melis GC, Boelens PG, Deutz NE, van Leeuwen PA, Dejong CHIntestinal and hepatic metabolism of glutamine and citrulline in humansJ Physiol.(2007 Jun 1)
- van de Poll MC, Siroen MP, van Leeuwen PA, Soeters PB, Melis GC, Boelens PG, Deutz NE, Dejong CHInterorgan amino acid exchange in humans: consequences for arginine and citrulline metabolismAm J Clin Nutr.(2007 Jan)
- Rougé C, Des Robert C, Robins A, Le Bacquer O, Volteau C, De La Cochetière MF, Darmaun DManipulation of citrulline availability in humansAm J Physiol Gastrointest Liver Physiol.(2007 Nov)
- Bommarius AS, Makryaleas K, Drauz KAn enzymatic route to L-ornithine from L-arginine–activation and stabilization studies on L-arginaseBiomed Biochim Acta.(1991)
- Bommarius AS, Drauz KAn enzymatic route to L-ornithine from arginine–activation, selectivity and stabilization of L-arginaseBioorg Med Chem.(1994 Jul)
- Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R, Servais A, Valayannopoulos V, Lindner M, Rubio V, Dionisi-Vici CSuggested guidelines for the diagnosis and management of urea cycle disordersOrphanet J Rare Dis.(2012 May 29)
- Targeted cellular metabolism for cancer chemotherapy with recombinant arginine-degrading enzymes.()
- Enzymes of Arginine Metabolism.()
- Lameu C, de Camargo AC, Faria ML-arginine signalling potential in the brain: the peripheral gets centralRecent Pat CNS Drug Discov.(2009 Jun)
- Flam BR, Eichler DC, Solomonson LPEndothelial nitric oxide production is tightly coupled to the citrulline-NO cycleNitric Oxide.(2007 Nov-Dec)
- Adverse Gastrointestinal Effects of Arginine and Related Amino Acids.()
- Morris SM JrRecent advances in arginine metabolismCurr Opin Clin Nutr Metab Care.(2004 Jan)
- De Bandt JP, Cynober L, Lim SK, Coudray-Lucas C, Poupon R, Giboudeau JMetabolism of ornithine, alpha-ketoglutarate and arginine in isolated perfused rat liverBr J Nutr.(1995 Feb)
- Curis E, Nicolis I, Moinard C, Osowska S, Zerrouk N, Bénazeth S, Cynober LAlmost all about citrulline in mammalsAmino Acids.(2005 Nov)
- Plasma arginine and citrulline kinetics in adults given adequate and arginine-free diets.
- Dai Z, Wu Z, Yang Y, Wang J, Satterfield MC, Meininger CJ, Bazer FW, Wu GNitric oxide and energy metabolism in mammalsBiofactors.(2013 Mar 29)
- Regunathan S, Reis DJCharacterization of arginine decarboxylase in rat brain and liver: distinction from ornithine decarboxylaseJ Neurochem.(2000 May)
- Raasch W, Regunathan S, Li G, Reis DJAgmatine, the bacterial amine, is widely distributed in mammalian tissuesLife Sci.(1995)
- Rougé C, Des Robert C, Robins A, Le Bacquer O, Volteau C, De La Cochetière MF, Darmaun DManipulation of citrulline availability in humansAm J Physiol Gastrointest Liver Physiol.(2007 Nov)
- Thibault R, Flet L, Vavasseur F, Lemerle M, Ferchaud-Roucher V, Picot D, Darmaun DOral citrulline does not affect whole body protein metabolism in healthy human volunteers: results of a prospective, randomized, double-blind, cross-over studyClin Nutr.(2011 Dec)
- Sureda A, Córdova A, Ferrer MD, Pérez G, Tur JA, Pons AL-citrulline-malate influence over branched chain amino acid utilization during exerciseEur J Appl Physiol.(2010 Sep)
- Sureda A, Cordova A, Ferrer MD, Tauler P, Perez G, Tur JA, Pons AEffects of L-citrulline oral supplementation on polymorphonuclear neutrophils oxidative burst and nitric oxide production after exerciseFree Radic Res.(2009 Sep)
- Stuehr DJStructure-function aspects in the nitric oxide synthasesAnnu Rev Pharmacol Toxicol.(1997)
- Charles IG, Scorer CA, Moro MA, Fernàndez C, Chubb A, Dawson J, Foxwell N, Knowles RG, Baylis SAExpression of human nitric oxide synthase isozymesMethods Enzymol.(1996)
- Casas JP, Cavalleri GL, Bautista LE, Smeeth L, Humphries SE, Hingorani ADEndothelial nitric oxide synthase gene polymorphisms and cardiovascular disease: a HuGE reviewAm J Epidemiol.(2006 Nov 15)
- Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ERLong-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthaseCell.(1996 Dec 13)
- Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MCEndothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOSScience.(1994 Jul 22)
- Griffith OW, Stuehr DJNitric oxide synthases: properties and catalytic mechanismAnnu Rev Physiol.(1995)
- 82.^Marletta MANitric oxide synthase structure and mechanismJ Biol Chem.(1993 Jun 15)
- Masters BSNitric oxide synthases: why so complexAnnu Rev Nutr.(1994)
- Mayer B, John M, Heinzel B, Werner ER, Wachter H, Schultz G, Böhme EBrain nitric oxide synthase is a biopterin- and flavin-containing multi-functional oxido-reductaseFEBS Lett.(1991 Aug 19)
- Hevel JM, White KA, Marletta MAPurification of the inducible murine macrophage nitric oxide synthase. Identification as a flavoproteinJ Biol Chem.(1991 Dec 5)
- Stuehr DJ, Cho HJ, Kwon NS, Weise MF, Nathan CFPurification and characterization of the cytokine-induced macrophage nitric oxide synthase: an FAD- and FMN-containing flavoproteinProc Natl Acad Sci U S A.(1991 Sep 1)
- Mayer B, Hemmens BBiosynthesis and action of nitric oxide in mammalian cellsTrends Biochem Sci.(1997 Dec)
- Ghosh DK, Abu-Soud HM, Stuehr DJReconstitution of the second step in NO synthesis using the isolated oxygenase and reductase domains of macrophage NO synthaseBiochemistry.(1995 Sep 12)
- Lekakis JP, Papathanassiou S, Papaioannou TG, Papamichael CM, Zakopoulos N, Kotsis V, Dagre AG, Stamatelopoulos K, Protogerou A, Stamatelopoulos SFOral L-arginine improves endothelial dysfunction in patients with essential hypertensionInt J Cardiol.(2002 Dec)
- L-Arginine and Atherothrombosis.()
- Lucotti P, Setola E, Monti LD, Galluccio E, Costa S, Sandoli EP, Fermo I, Rabaiotti G, Gatti R, Piatti PBeneficial effects of a long-term oral L-arginine treatment added to a hypocaloric diet and exercise training program in obese, insulin-resistant type 2 diabetic patientsAm J Physiol Endocrinol Metab.(2006 Nov)
- Acute L-arginine supplementation reduces the O2 cost of moderate-intensity exercise and enhances high-intensity exercise tolerance.()
- Fahs CA, Heffernan KS, Fernhall BHemodynamic and vascular response to resistance exercise with L-arginineMed Sci Sports Exerc.(2009 Apr)
- Tang JE, Lysecki PJ, Manolakos JJ, MacDonald MJ, Tarnopolsky MA, Phillips SMBolus arginine supplementation affects neither muscle blood flow nor muscle protein synthesis in young men at rest or after resistance exerciseJ Nutr.(2011 Feb)
- Alvares TS, Conte CA, Paschoalin VM, Silva JT, Meirelles Cde M, Bhambhani YN, Gomes PSAcute l-arginine supplementation increases muscle blood volume but not strength performanceAppl Physiol Nutr Metab.(2012 Feb)
- Durante W, Johnson FK, Johnson RAArginase: a critical regulator of nitric oxide synthesis and vascular functionClin Exp Pharmacol Physiol.(2007 Sep)
- Baydoun AR, Emery PW, Pearson JD, Mann GESubstrate-dependent regulation of intracellular amino acid concentrations in cultured bovine aortic endothelial cellsBiochem Biophys Res Commun.(1990 Dec 31)
- Palmer RM, Ashton DS, Moncada SVascular endothelial cells synthesize nitric oxide from L-arginineNature.(1988 Jun 16)
- Cardounel AJ, Cui H, Samouilov A, Johnson W, Kearns P, Tsai AL, Berka V, Zweier JLEvidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular functionJ Biol Chem.(2007 Jan 12)
- Hardy TA, May JMCoordinate regulation of L-arginine uptake and nitric oxide synthase activity in cultured endothelial cellsFree Radic Biol Med.(2002 Jan 15)
- Bode-Böger SM, Scalera F, Ignarro LJThe L-arginine paradox: Importance of the L-arginine/asymmetrical dimethylarginine ratioPharmacol Ther.(2007 Jun)
- Shin S, Mohan S, Fung HLIntracellular L-arginine concentration does not determine NO production in endothelial cells: implications on the “L-arginine paradox”Biochem Biophys Res Commun.(2011 Nov 4)
- Liu TH, Wu CL, Chiang CW, Lo YW, Tseng HF, Chang CKNo effect of short-term arginine supplementation on nitric oxide production, metabolism and performance in intermittent exercise in athletesJ Nutr Biochem.(2009 Jun)
- Tsukahara H, Gordienko DV, Goligorsky MSContinuous monitoring of nitric oxide release from human umbilical vein endothelial cellsBiochem Biophys Res Commun.(1993 Jun 15)
- Joshi MS, Ferguson TB Jr, Johnson FK, Johnson RA, Parthasarathy S, Lancaster JR JrReceptor-mediated activation of nitric oxide synthesis by arginine in endothelial cellsProc Natl Acad Sci U S A.(2007 Jun 12)
- McDonald KK, Zharikov S, Block ER, Kilberg MSA caveolar complex between the cationic amino acid transporter 1 and endothelial nitric-oxide synthase may explain the “arginine paradox”J Biol Chem.(1997 Dec 12)
- Zani BG, Bohlen HGTransport of extracellular l-arginine via cationic amino acid transporter is required during in vivo endothelial nitric oxide productionAm J Physiol Heart Circ Physiol.(2005 Oct)
- Kone BCProtein-protein interactions controlling nitric oxide synthasesActa Physiol Scand.(2000 Jan)
- Vallance P, Leone A, Calver A, Collier J, Moncada SAccumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failureLancet.(1992 Mar 7)
- Witte DR, van der Graaf Y, Grobbee DE, Bots ML; SMART Study GroupMeasurement of flow-mediated dilatation of the brachial artery is affected by local elastic vessel wall properties in high-risk patientsAtherosclerosis.(2005 Oct)
- Lind LArterial compliance influences the measurement of flow-mediated vasodilation, but not acetylcholine-mediated forearm blood flow. The Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) studyAtherosclerosis.(2007 Jan)
- Zajac A, Poprzecki S, Zebrowska A, Chalimoniuk M, Langfort JArginine and ornithine supplementation increases growth hormone and insulin-like growth factor-1 serum levels after heavy-resistance exercise in strength-trained athletesJ Strength Cond Res.(2010 Apr)
- Kanaley JAGrowth hormone, arginine and exerciseCurr Opin Clin Nutr Metab Care.(2008 Jan)
- Oral arginine attenuates the growth hormone response to resistance exercise.()
- Marcell TJ, Taaffe DR, Hawkins SA, Tarpenning KM, Pyka G, Kohlmeier L, Wiswell RA, Marcus ROral arginine does not stimulate basal or augment exercise-induced GH secretion in either young or old adultsJ Gerontol A Biol Sci Med Sci.(1999 Aug)
- Borst SE, Millard WJ, Lowenthal DTGrowth hormone, exercise, and aging: the future of therapy for the frail elderlyJ Am Geriatr Soc.(1994 May)
- Fogelholm GM, Näveri HK, Kiilavuori KT, Härkönen MHLow-dose amino acid supplementation: no effects on serum human growth hormone and insulin in male weightliftersInt J Sport Nutr.(1993 Sep)
- Veldhuis JD, Bowers CYRegulated recovery of pulsatile growth hormone secretion from negative feedback: a preclinical investigationAm J Physiol Regul Integr Comp Physiol.(2011 Oct)
- Veldhuis JD, Anderson SM, Shah N, Bray M, Vick T, Gentili A, Mulligan T, Johnson ML, Weltman A, Evans WS, Iranmanesh ANeurophysiological regulation and target-tissue impact of the pulsatile mode of growth hormone secretion in the humanGrowth Horm IGF Res.(2001 Jun)
- Besset A, Bonardet A, Rondouin G, Descomps B, Passouant PIncrease in sleep related GH and Prl secretion after chronic arginine aspartate administration in manActa Endocrinol (Copenh).(1982 Jan)
- Maxwell AJ, Ho HV, Le CQ, Lin PS, Bernstein D, Cooke JPL-arginine enhances aerobic exercise capacity in association with augmented nitric oxide productionJ Appl Physiol.(2001 Mar)
- Schaefer A, Piquard F, Geny B, Doutreleau S, Lampert E, Mettauer B, Lonsdorfer JL-arginine reduces exercise-induced increase in plasma lactate and ammoniaInt J Sports Med.(2002 Aug)
- Acute L-arginine supplementation reduces the O2 cost of moderate-intensity exercise and enhances high-intensity exercise tolerance.
- Wax B, Kavazis AN, Webb HE, Brown SPAcute L-arginine alpha ketoglutarate supplementation fails to improve muscular performance in resistance trained and untrained menJ Int Soc Sports Nutr.(2012 Apr 17)
- Buford BN, Koch AJGlycine-arginine-alpha-ketoisocaproic acid improves performance of repeated cycling sprintsMed Sci Sports Exerc.(2004 Apr)
- bel T, Knechtle B, Perret C, Eser P, von Arx P, Knecht HInfluence of chronic supplementation of arginine aspartate in endurance athletes on performance and substrate metabolism – a randomized, double-blind, placebo-controlled studyInt J Sports Med.(2005 Jun)
- Colombani PC, Bitzi R, Frey-Rindova P, Frey W, Arnold M, Langhans W, Wenk CChronic arginine aspartate supplementation in runners reduces total plasma amino acid level at rest and during a marathon runEur J Nutr.(1999 Dec)
- McConell GKEffects of L-arginine supplementation on exercise metabolismCurr Opin Clin Nutr Metab Care.(2007 Jan)
- Moinard C, Nicolis I, Neveux N, Darquy S, Bénazeth S, Cynober LDose-ranging effects of citrulline administration on plasma amino acids and hormonal patterns in healthy subjects: the Citrudose pharmacokinetic studyBr J Nutr.(2008 Apr)
- Rougé C, Des Robert C, Robins A, Le Bacquer O, Volteau C, De La Cochetière MF, Darmaun DManipulation of citrulline availability in humansAm J Physiol Gastrointest Liver Physiol.(2007 Nov)
- Thibault R, Flet L, Vavasseur F, Lemerle M, Ferchaud-Roucher V, Picot D, Darmaun DOral citrulline does not affect whole body protein metabolism in healthy human volunteers: results of a prospective, randomized, double-blind, cross-over studyClin Nutr.(2011 Dec)
- Sureda A, Córdova A, Ferrer MD, Pérez G, Tur JA, Pons AL-citrulline-malate influence over branched chain amino acid utilization during exerciseEur J Appl Physiol.(2010 Sep)
- https://pubmed.ncbi.nlm.nih.gov/34417881/
- Valenzuela PL, Morales JS, Emanuele E, Pareja-Galeano H, Lucia ASupplements with purported effects on muscle mass and strength.Eur J Nutr.(2019-Dec)
- The role of glutamine and α-ketoglutarate in gut metabolism and the potential application in medicine and nutritionJ Pre Clin Clin Res.(2007-01)
- Bill Campbell, Mike Roberts, Chad Kerksick, Colin Wilborn, Brandon Marcello, Lem Taylor, Erika Nassar, Brian Leutholtz, Rodney Bowden, Chris Rasmussen, Mike Greenwood, Richard KreiderPharmacokinetics, safety, and effects on exercise performance of L-arginine alpha-ketoglutarate in trained adult menNutrition.(2006 Sep)
- Wu N, Yang M, Gaur U, Xu H, Yao Y, Li DAlpha-Ketoglutarate: Physiological Functions and Applications.Biomol Ther (Seoul).(2016-Jan)
- Bayliak MM, Lushchak VIPleiotropic effects of alpha-ketoglutarate as a potential anti-ageing agent.Ageing Res Rev.(2021-Mar)
- Azar Asadi Shahmirzadi, Daniel Edgar, Chen-Yu Liao, Yueh-Mei Hsu, Mark Lucanic, Arash Asadi Shahmirzadi, Christopher D Wiley, Garbo Gan, Dong Eun Kim, Herbert G Kasler, Chisaka Kuehnemann, Brian Kaplowitz, Dipa Bhaumik, Rebeccah R Riley, Brian K Kennedy, Gordon J LithgowAlpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging MiceCell Metab.(2020 Sep 1)
- Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, Jung G, Solis GM, Fazlollahi F, Kaweeteerawat C, Quach A, Nili M, Krall AS, Godwin HA, Chang HR, Faull KF, Guo F, Jiang M, Trauger SA, Saghatelian A, Braas D, Christofk HR, Clarke CF, Teitell MA, Petrascheck M, Reue K, Jung ME, Frand AR, Huang JThe metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR.Nature.(2014-Jun-19)
- Guo L, Chen S, Ou L, Li S, Ye ZN, Liu HFDisrupted Alpha-Ketoglutarate Homeostasis: Understanding Kidney Diseases from the View of Metabolism and Beyond.Diabetes Metab Syndr Obes.(2022)
- Riedel E, Hampl H, Steudle V, Nündel MCalcium alpha-ketoglutarate administration to malnourished hemodialysis patients improves plasma arginine concentrations.Miner Electrolyte Metab.(1996)
- Xue H, Tu Y, Zhang G, Xin X, Hu H, Qiu W, Ruan D, Zhao YMechanism of ultrasound and tea polyphenol assisted ultrasound modification of egg white protein gel.Ultrason Sonochem.(2021-Dec)
- Son ED, Choi GH, Kim H, Lee B, Chang IS, Hwang JSAlpha-ketoglutarate stimulates procollagen production in cultured human dermal fibroblasts, and decreases UVB-induced wrinkle formation following topical application on the dorsal skin of hairless mice.Biol Pharm Bull.(2007-Aug)
- Gyanwali B, Lim ZX, Soh J, Lim C, Guan SP, Goh J, Maier AB, Kennedy BKAlpha-Ketoglutarate dietary supplementation to improve health in humans.Trends Endocrinol Metab.(2022-Feb)
- Naeini SH, Mavaddatiyan L, Kalkhoran ZR, Taherkhani S, Talkhabi MAlpha-ketoglutarate as a potent regulator for lifespan and healthspan: Evidences and perspectives.Exp Gerontol.(2023-May)
- Shao A, Hathcock JNRisk assessment for the amino acids taurine, L-glutamine and L-arginineRegul Toxicol Pharmacol.(2008 Apr)
- Hellier MD, Holdsworth CD, Perrett DDibasic amino acid absorption in manGastroenterology.(1973 Oct)
- Izzo AA, Mascolo N, Capasso FNitric oxide as a modulator of intestinal water and electrolyte transportDig Dis Sci.(1998 Aug)