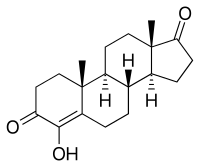
Quando si è sottoposti a stress fisico e psicologico il corpo produce Cortisolo extra, il che fa più male che bene. Su questo, nessuna novità. Secondo uno studio giapponese svolto su animali, pubblicato nel Journal of Clinical Biochemistry, e dai dati del Dutch National Institute for Public Health and the Environment (RIVM), esiste un modo per ridurre il livello di Cortisolo, semplicemente aumentando l’assunzione di vitamina E.(1)
I ricercatori hanno …fatto esperimenti sui ratti. Alcuni dei ratti erano giovani; Altri erano di età avanzata. Ad alcuni ratti è stato somministrato cibo contenente pochissima vitamina E e alcuni ratti sono stati messi in una gabbia in cui l’aria era composta interamente di ossigeno. L’età avanzata, una carenza di vitamina E e un’atmosfera ricca di ossigeno sono tre fattori che aumentano l’attività dei radicali liberi: molecole aggressive che causano danni alle cellule. In questo articolo consideriamo l’effetto di una carenza di vitamina E.
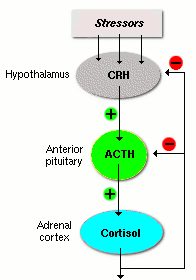
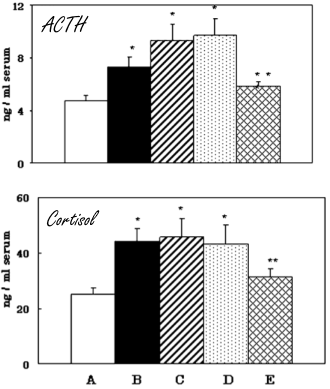
Nei giovani ratti che sono stati sottoposti a una carenza di vitamina E [D], dopo poche settimane il loro ipotalamo ha iniziato a produrre più CRH, l’ipofisi più ACTH e le ghiandole surrenali più Cortisolo rispetto ai giovani ratti che sono ai quali era stata data una normale e dieta equilibrata [A].
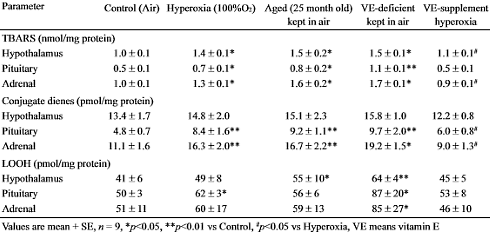
Una carenza di vitamina E ha aumentato la concentrazione di TBARS e altri marker dell’attività dei radicali liberi nell’ipotalamo, nell’ipofisi e nelle ghiandole surrenali dei ratti. A quanto pare l’attività degli agenti ossidanti aumenta la produzione di Cortisolo.
Lo studio giapponese svolto su animali è rilevante per gli esseri umani, in particolare per le donne. Secondo le cifre raccolte dal RIVM sull’assunzione di cibo, circa il 50-60% delle donne olandesi e il 20-30% degli uomini olandesi consumano meno vitamina E rispetto alla quantità di cui hanno bisogno.
Gli adulti hanno bisogno di circa 10 mg di vitamina E al giorno. Questa quantità si trova in una manciata generosa di mandorle. Un avocado o una porzione di spinaci cotti contengono 4mg di vitamina E. Un kiwi contiene 1 mg di vitamina E.
Come riportato dai ricercatori, i risultati ottenuti in questo studio suggeriscono che lo stress ossidativo induca danni ossidativi nell’ippocampo, con conseguente iper secrezione del corticosteroide. È anche chiaro che la vitamina E impedisce il verificarsi di questo fenomeno attraverso le sue proprietà antiossidanti.
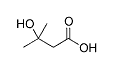
Il Formestano (nome commerciale Lentaron), noto anche come 4-idrossi-andro-4-eno-3,17-dione, è un inibitore selettivo dell’aromatasi di tipo I, steroideo. (1) Questo agente è strutturalmente un derivato del Androstenedione, differendo da questo ben noto pro-ormone dall’aggiunta di un gruppo 4-idrossile. Questa aggiunta, tuttavia, è responsabile del legame irreversibile che si crea tra il Formestano e l’enzima aromatasi. Ciò significa che il Formestano si lega con l’enzima aromatasi rendendo quest’ultimo definitivamente inattivo. L’enzima dovrà essere sostituito, attraverso il normale metabolismo, prima che il corpo sia in grado di recuperare la sua piena capacità di sintesi estrogenica tramite l’azione dell’enzima aromatasi. Ciò può richiedere alcuni giorni o più dopo la cessazione della terapia. Data questa modalità d’azione, il Formestano è appunto definito come inibitore dell’aromatasi “suicida”, in quanto il farmaco essenzialmente si “sacrifica” nel processo di blocco della conversione degli androgeni in estrogeni. Come ben sappiamo, questa classe di farmaci, gli inibitori dell’aromatasi, sono utilizzati (off-label) principalmente dagli atleti di sesso maschile per prevenire gli effetti collaterali estrogenici dati dagli AAS soggetti all’aromatizzazione e, soprattutto per le atlete, per aumentare la perdita di grasso e la definizione muscolare durante la dieta.
A causa della sua potente azione anti-aromatasi, il Formestano è stato usato clinicamente per curare i pazienti affetti da tumore al seno in diversi paesi, tra cui Inghilterra, Germania, Svizzera, Spagna, Australia, Nuova Zelanda, Italia e Malesia. È stato dimostrato essere un’opzione efficace come seconda linea di difesa dopo il Tamoxifene, un antagonista del recettore degli estrogeni, non avendo potuto provocare una risposta positiva nei pazienti producendo una risposta complessiva statisticamente simile a quella del Tamoxifene quando somministrato come terapia di prima linea. In termini di potenza complessiva, il Formestano non è più forte degli inibitori dell’aromatasi di terza generazione come Arimidex (Anastrozolo) o Femara (Letrozolo). In uno studio, ad esempio, è stato osservato un livello di soppressione dei livelli estrogenici del 79% in seguito alla somministrazione di 1mg di Arimidex al giorno per 4 settimane (pari a livelli noti con l’uso di Femara), ma solo un livello di soppressione del 58% con iniezioni intramuscolari di Formestano (250 Mg ogni due settimane). (2) Ma in confronto ai Modulatori Selettivi del Recettore degli Estrogeni (SERM) come il Nolvadex (Tamoxifene citrato), il Formestano è significativamente più efficace nel controllo estrogenico.
Studi hanno mostrato una diminuzione delle SHBG fino al 34% in seguito all’uso di Formestano, (3) il che porta ad una maggiore frazione libera degli AAS soggetti al legame con le proteine di trasporto ( vedi, ad esempio, Testosterone e Boldenone).
Il Formestano è stato il primo inibitore selettivo dell’aromatasi ad essere sviluppato come farmaco da prescrizione, e apparve in Europa alla metà degli anni ’90 sotto il nome commerciale di Lentaron Depot. È stato venduto dalla Novartis, che commercializza Lentaron Depot in due dozzine di paesi tra cui Argentina, Austria, Belgio, Brasile, Canada, Cile, Repubblica Ceca, Danimarca, Francia, Germania, Grecia, Hong Kong, Irlanda, Israele, Paesi Bassi, Portogallo, Sudafrica, Spagna, Svizzera, Turchia e Regno Unito. Con la comparsa degli inibitori dell’aromatasi più recenti e più efficaci, il Formestano ha subito perso la sua presenza nel mercato ad un ritmo rapido. La maggior parte dei preparati iniziali di Lentaron Depot hanno subito un interruzione di produzione. Il farmaco rimane tuttavia disponibile oggi, ma solo in un piccolo numero di nazioni (Austria, Brasile, Repubblica Ceca, Hong Kong e Turchia).
Il Formestano viene usato nel trattamento del cancro al seno recettore-positivo dell’estrogeno nelle donne in postmenopausa. Il dosaggio terapeutico consigliato è di 250 mg iniettati intramuscolarmente ogni due settimane. Sebbene non sia una forma medica approvata del farmaco, gli studi hanno dimostrato che un livello di soppressione estrogenica simile a quello raggiunto con somministrazione tramite iniezione intramuscolare può essere raggiunto anche con l’uso orale di Formestano. A causa della scarsa biodisponibilità, tuttavia, la dose orale necessaria è di circa 250mg al giorno. Quando usato (off-label) per mitigare gli effetti collaterali estrogenici derivati dall’uso di AAS aromatizzabili o per aumentare la definizione muscolare, gli atleti di sesso maschile spesso assumono il farmaco tramite iniezioni intramuscolari da 250mg ogni due settimane o per via orale a 250 mg al giorno. Esistono anche prodotti topici contenenti Formestano venduti nei siti di integratori UK. In questo caso, questi prodotti contengono mediamente 25mg di Formestano per ml, e i produttori riportano in etichetta modalità di applicazione solitamente di 1ml per 2-3 volte al giorno.
L’emivita del Formestano è indicata essere di circa 4 giorni.
Gli effetti collaterali comuni associati all’uso di un inibitore dell’aromatasi includono vampate di calore, dolore alle articolazioni, debolezza, affaticamento, alterazioni dell’umore, depressione, pressione alta, gonfiore a braccia / gambe e mal di testa. Gli inibitori dell’aromatasi possono anche diminuire la densità minerale ossea, che può portare ad osteoporosi e ad un aumento delle fratture nei pazienti sensibili. Alcuni individui possono anche rispondere al farmaco con effetti collaterali gastrointestinali tra cui nausea e vomito. Gli inibitori dell’aromatasi possono danneggiare lo sviluppo del feto e non devono mai essere assunti durante la gravidanza. Quando assunti dagli uomini (off label) per ridurre i livelli di estrogeni circolanti durante cicli di AAS aromatizzabili prolungati, gli inibitori dell’aromatasi possono aumentare il rischio di malattia cardiovascolare (CVD) inficiando alcune proprietà benefiche degli estrogeni sui valori del colesterolo. Gli studi hanno dimostrato che quando un AAS aromatizzabile come il Testosterone Enantato viene assunto in combinazione con un inibitore dell’aromatasi, la soppressione dei livelli di colesterolo HDL (buono) diventa significativamente più pronunciata. Poiché i SERM (come il Nolvadex®) non abbassano i livelli circolanti degli estrogeni ma ne impediscono l’attività recettoriale non presentano generalmente lo stesso effetto negativo sui valori di colesterolo.
Il Formestano non è ampiamente disponibile come farmaco da prescrizione, e di conseguenza non è molto diffuso tra gli atleti. Come detto in precedenza, sono presenti prodotti contenenti Formestano (in forma per somministrazione topica) venduti in siti di integratori UK.
Gabriel Bellizzi
Riferimenti:
1- Pérez Carrión R, Alberola Candel V, Calabresi F, et al. (1994). “Comparison of the selective aromatase inhibitor formestane with tamoxifen as first-line hormonal therapy in postmenopausal women with advanced breast cancer”. Ann. Oncol. 5 Suppl 7: S19–24. PMID7873457
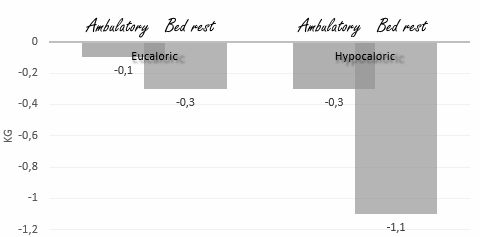
Qualche tempo fa ho riportato che un surplus calorico durante un periodo di inattività fisica accelera la perdita di massa muscolare. Questa scoperta ha spinto a speculare che durante un periodo di inattività, un lieve deficit calorico potrebbe essere il modo per tenere sotto controllo questo effetto. Ma se questo fosse il caso, allora il deficit calorico dovrebbe essere davvero modesto, e lo possiamo dedurre… leggendo lo studio che i ricercatori italiani dell’Università di Trieste hanno pubblicato nel 2007 sul American Journal of Clinical Nutrition.(1) Un deficit calorico del 20% in ogni caso è troppo grande: questo stimola solo la perdita di massa muscolare.
I ricercatori hanno reclutato per l’esperimento 9 partecipanti sani facendoli stare a riposo per 14 giorni [Bed rest] in due occasioni separate. In una occasione i partecipanti hanno consumato una quantità calorica pari al loro fabisogno giornaliero [Eucaloric]; in un altra occasione hanno consumato una quantità calorica inferiore al 20% del loro fabisogno giornaliero [Hypocaloric]. I ricercatori hanno misurato l’effetto di questo trattamento sulla composizione corporea dei partecipanti.
In altre due occasioni i partecipanti sono stati autorizzati a camminare [Ambulatory]; durante una di queste occasioni della durata di 14 giorni hai partecipanti è stato fornito un quantitativo calorico pari al loro dispendio giornaliero e in un’altra il 20% in meno delle calorie consumate.
I soggetti a riposo [Bed rest] hanno mostrato una perdita della massa corporea magra – e l’ammontare della perdita era quattro volte maggiore quando i partecipanti seguivano una dieta ipocalorica.
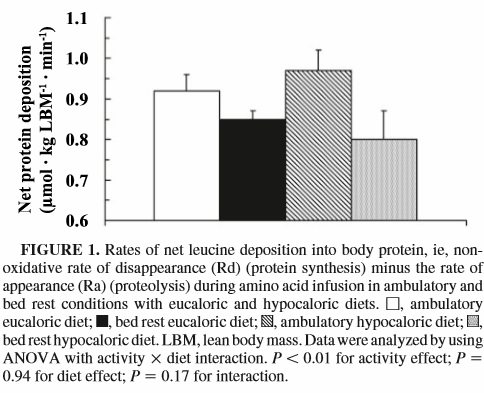
Dalla figura riportata qui sopra si può vedere che durante l’inattività i muscoli assorbono meno leucina dal flusso ematico riducendo anche gli effetti anabolizzanti indotti dalla leucina. Quindi, l’inattività fisica sembra ridurre i processi anabolizzanti nelle cellule muscolari. E un deficit calorico rafforza questa riduzione.
La Fentermina [2-methyl-1- phenylpropan-2-amine (2-methyl-amphetamine)],(fenil-terziario-butilammina), nota anche come α,α-dimethylphenethylamine, commercializzata come Fentermina cloridrato, è uno stimolante simpaticomimetico della famiglia delle anfetamine. Come altri derivati delle amfetamine, viene classificato come agente anoressizzante (soppressore dell’appetito). La Fentermina è comunemente prescritta come coadiuvante per la perdita di peso in pazienti obesi purché in associazione con esercizio fisico e una dieta dimagrante. Viene di solito utilizzata per brevi periodi di tempo (meno di 12 settimane). L’obiettivo principale del farmaco è quello di ridurre il desiderio di mangiare, riducendo così l’assunzione calorica totale. Anche se i dati sembrano variare da uno studio all’altro, gran parte di essi supportano almeno una modesta perdita aggiuntiva di massa grassa con l’uso di Fentermina cloridrato. (1) Gli atleti usano la Fentermina cloridrato per lo stesso scopo, in genere durante i periodi di forte restrizione calorica.
Nel 1959, la Fentermina base ricevette l’approvazione da parte della FDA degli Stati Uniti come farmaco anti-appetito. (2) Alla fine divenne disponibile in forma di sale cloridrato e in forma di resina. (2) La Fentermina cloridrato è stata introdotto per la prima volta nel mercato degli Stati Uniti negli anni ’70. La Fentermina era da tempo utilizzata come soppressore dell’appetito, anche se godette di maggiore attenzione nei primi anni ’90, quando il farmaco è stato associato con successo alla Fenfluramina durante studi dietetici. I ricercatori hanno dimostrato che questo tipo di combinazione di farmaci era effettivamente più efficace nel promuovere la perdita di peso rispetto alla sola dieta ed esercizio fisico, risultati che hanno portato rapidamente il Fen-Phen ad essere di primo piano nel mercato dei farmaci da prescrizione per la perdita di peso. Nel 1997, tuttavia, emerse che una percentuale molto elevata di utilizzatori di Fen-Phen aveva riportato difetti alla valvola cardiaca a causa dei farmaci. La Fenfluramina è stata identificata come causa principale di tale evento, e venne ritirata dal mercato USA nello stesso anno. La Fentermina oggi rimane disponibile negli Stati Uniti e in molte altre nazioni. I nomi commerciali più popolari sotto i quali viene venduta la Fentermina cloridrato includono Adipex, Ionamin, Anoxine, Phentrol e Obenix. Si noti che come derivato delle anfetamine, questo farmaco ha la tendenza a portare ad assuefazione e dipendenza. Per questo motivo è stato aggiunto alla lista IV delle sostanze controllate negli Stati Uniti.
La Fentermina ha qualche somiglianza nella sua farmacodinamica con il suo composto madre, l’anfetamina, in quanto entrambe agonisti TAAR1 (3), dove l’attivazione di questi nei neuroni monoammina facilita l’efflusso o il rilascio nella sinapsi di questi neurochimici; A dosi clinicamente rilevanti, la Fentermina agisce principalmente come agente di rilascio della norepinefrina nei neuroni, anche se, in misura minore, rilascia anche la dopamina e la serotonina nelle sinapsi. (4)(5) La Fentermina può anche innescare il rilascio di monoammine dal VMAT2, che è un effetto farmacodinamico comune tra i sostituti delle anfetamine. Il meccanismo d’azione principale della Fentermina nel trattamento dell’obesità è la riduzione della percezione della fame, che è un processo cognitivo mediato principalmente attraverso diversi nuclei all’interno dell’ipotalamo (in particolare il nucleo ipotalamico laterale, il nucleo arcuato e il nucleo ventromediale). Al di fuori del cervello, la Fentermina rilascia norepinefrina e epinefrina – note anche come noradrenalina e adrenalina – causando il rilascio di acidi grassi da parte delle cellule adipose.
Per un’efficacia ottimale, la Fentermina cloridrato non deve essere assunta con il cibo. La dose per i soggetti adulti è una capsula o compressa (37,5 mg) al giorno, somministrata prima o dopo 1-2 ore dalla prima colazione. Per alcuni pazienti mezza compressa (18,75 mg) al giorno può essere sufficiente, mentre in altri casi può essere consigliabile la somministrazione di mezza compressa (18,75 mg) due volte al giorno. Se assunta più di una volta al giorno, la seconda dose non deve mai essere somministrata nelle 4-6 ore precedenti all’andare a dormire. Il farmaco viene utilizzato tipicamente per 3-4 settimane alla volta, con terapia raramente superiori alle 12 settimane. Gli atleti usano tipicamente la quantità da prescrizione e a breve termine, a causa della elevata probabilità di effetti collaterali, poiché la possibilità che ciò si verifichi è elevata oltre i normali tempi terapeutici.
La Fentermina cloridrato è disponibile in diversi Paesi. Non è ampiamente contraffatta. In Italia non è in commercio pertanto è vietata l’importazione anche per uso personale.
5- Rothman RB, Baumann MH, Dersch CM, et al. (January 2001). “Amphetamine-type central nervous system stimulants release norepinephrine more potently than they dopamine and serotonin”. Synapse. 39 (1): 32–41. doi:10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. PMID11071707.
La supplementazione con la forma libera di HMB potrebbe forse offrire una certa protezione contro la cirrosi epatica, il danno renale, l’insufficienza cardiaca e la demenza. Ciò si deduce da uno studio svolto su esseri umani che gli scienziati dello sport della University of Central Florida hanno riportato sul Growth Hormone & IGF Research. (1) Secondo lo studio, l’HMB riduce la concentrazione della proteina di trasporto IGFBP-7 nel sangue. Ma questa riduzione è sufficiente per avere un effetto? È probabile che sia estremamente modesto …
Studio
I ricercatori hanno somministrato ad una dozzina di soldati addestrati d’elite 3g di HMB Free Acid ogni giorno per 23 giorni e hanno somministrato ad un’altra dozzina di soldati un placebo. Alla fine del periodo di integrazione i soldati hanno svolto una pesante sessione di addestramento, completa di privazione del sonno, marcia con attrezzature e senza dubbio un sergente capace di produrre una rispettabile quantità di decibel. I ricercatori hanno analizzato il sangue dei soldati prima e dopo l’integrazione.
Risultato
L’integrazione di HMB non ha avuto alcun effetto sulla concentrazione di IGF-1 nel sangue dei soldati. Né ha avuto un effetto sulla concentrazione delle proteine leganti l’IGF, IGFBP-1, IGFBP-2, IGFBP-3, IGFBP-4, IGFBP-5 e IGFBP-6. Ma l’HMB ha comportato una riduzione modesta ma statisticamente significativa delle concentrazioni di IGFBP-7.
I ricercatori non hanno potuto dare un’indicazione del effetto sulla salute di una diminuzione del livello IGFBP-7. Nulla è noto sul ruolo del IGFBP-7 nello stress, o nell’allenamento e il danno muscolare.
Conclusione
I ricercatori hanno scritto che, sebbene i risultati di questo studio non supportino l’influenza dell’HMB supplementare sulle concentrazioni di IGF-1 o IGFBPs-1-6 durante l’allenamento militare ad alta intensità, essa presenta le prove iniziali di una sua possibile azione sulla riduzione delle concentrazioni circolanti di IGFBP-7. Ciò può fornire qualche indicazione di una ridotta risposta allo stress, ma è necessaria un’ulteriore ricerca sul ruolo fisiologico del IGFBP-7 e l’addestramento militare.
Maggiori informazioni su IGFB-7
L’IGFBP-7 sembra accelerare la cirrosi epatica (2), e ciò rende le sostanze potenzialmente in grado di ridurre i livelli di IGFBP-7 di interesse per i pazienti la cui malattia è in una fase avanzata. Le stesse sostanze possono essere utili anche per i cardiologi, nefrologi, diabetologi e neurologi. I cardiologi osservano una ridotta insufficienza cardiaca e ipertrofia cardiaca nelle persone con livelli ematici di IGFBP-7 relativamente bassi. (3)(4) I nefrologi considerano un’alta concentrazione di IGFBP-7 nel sangue come un indicatore del danno renale. (5) Negli studi epidemiologici è stata dimostrata una bassa concentrazione di IGFBP-7 nel sangue che offre una protezione contro il diabete di tipo 2 (6), e studi su animali hanno suggerito ai neurologi che gli inibitori del IGFBP-7 possono aiutare a prevenire o addirittura a curare la demenza.(7)
Una riduzione della concentrazione di IGFBP-7 può comunque avere effetti meno attraenti. L’IGFBP-7 inibisce le cellule tumorali del seno (8), e nei moscerini della frutta la secrezione di IGFBP-7 da parte dei muscoli ha un effetto età-ritardante.(9)
Tutto ciò è interessante, ma ci si chiede se le persone che usano l’HMB dovrebbero prendere in considerazione tutte le ricerche. Gli effetti del HMB sulla concentrazione di IGFBP-7 sono davvero molto modeste.
Sponsor
Lo studio è stato finanziato dalla Metabolic Technologies. Questa è la compagnia dei ricercatori che hanno scoperto gli effetti anabolizzanti dell’HMB. La maggior parte dei brevetti per l’utilizzo del HMB sono archiviati sotto il nome di Metabolic Technologies.
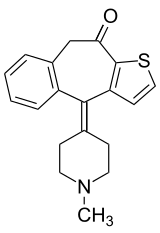
Il Ketotifene è un farmaco antistaminico di seconda generazione, antagonista non competitivo dei recettori H1 dell’istamina. Il farmaco si trova in commercio come un sale dell’acido fumarico, Ketotifene fumarato [4- (1-Metil-4-piperidilidene-4Hbenzo[4,5] cicloespta [1,2-b] tifene-1- (9H) -one fumarato.], ed è disponibile sia nella forma di soluzione oftalmica allo 0.025% che di compresse da 2 mg. Il Ketotifene è venduto in Italia dalla società farmaceutica Thèa Farma con il nome di Zaditen. Poiché il brevetto è scaduto diverse società farmaceutiche commercializzano la molecola come medicinale equivalente.
Il Ketotifene è stato reso un farmaco da prescrizione a livello globale da parte della Novartis. È attualmente prescritto per le allergie, condizioni allergiche e (più comunemente) la gestione dell’asma in più di tre dozzine di paesi del mondo. Il nome commerciale più ampiamente disponibile è Zaditen (Novartis), che viene venduto in gran parte d’Europa e Asia. Oltre alle forme generiche del farmaco, decine di altri nomi commerciali possono essere trovati in mercati diversi. Il Ketotifene fumarato è approvato per la vendita negli Stati Uniti, ma attualmente solo come soluzione antiallergica oftalmica (Zaditor), non come farmaco orale per il trattamento di allergie o asma. Il dosaggio di Ketotifene fumarato in questo prodotto è troppo basso per essere considerato utile per qualsiasi altro scopo (off label). Data la pronta disponibilità di Ketotifene fumarato in altri paesi, il farmaco viene facilmente deviato per la vendita nel mercato nero. Comunque sia, non è estremamente popolare tra gli atleti.
Le linee guida UK sulla gestione clinica dell’asma considerano il Ketotifene un composto inefficace per la gestione di questa malattia. I dati disponibili sulla potenziale utilità del Ketotifene fumarato a tal fine sono conflittuali, con alcuni studi che riportano risultati positivi e altri che mostrano un effetto insignificante. Una revisione approfondita dei dati pubblicati sul Cochrane Database delle revisioni sistematiche del 2004 ha concluso che la molecola sembrava avere qualche utilità nel controllare l’asma e il respiro affannoso in molti bambini, ma la variabilità della malattia e della risposta al farmaco evidenzia che questi risultati positivi potrebbero non essere generalizzati per tutti i pazienti con asma. (1)
Il Ketotifene fumarato aumenta la concentrazione dei recettori beta-adrenergici nel corpo (in particolare i recettori beta-2). Farmaci che stimolano i recettori beta-2 sono comunemente prescritti come broncodilatatori, usati per aumentare il flusso d’aria nei polmoni e contrastare la costrizione causata dall’asma. Sebbene potenzialmente efficaci da soli, un effetto terapeutico chiave del Ketotifene fumarato è quello di aumentare la sensibilità del corpo ai farmaci beta-agonisti.
La sovraregolazione dei recettori beta-2 ad opera del Ketotifene fumarato rende questo farmaco interessante per gli atleti. Ciò è dovuto al forte ruolo del recettore beta-2 nel supportare la perdita di grasso. Anche se non è un forte composto per la perdita di grasso assunto da solo, in combinazione con un beta-2 agonista come il Clenbuterolo, il Ketotifene fumarato può aumentarne la potenza termogenica e prolungare notevolmente la finestra della lipolisi attiva. Il Clenbuterolo e gli altri beta-2 agonisti hanno normalmente una durata limitata di utilità in quanto i recettori beta-2-adrenergici diminuiscono di numero con una stimolazione regolare. Dopo un paio di settimane dall’inizio della terapia con questi composti, in genere cominciano a diminuire di efficacia. Il Ketotifene può prolungare notevolmente questo periodo di tempo.
La capacità del Ketotifene di potenziare gli effetti dei farmaci beta-2 agonisti è stata dimostrata in numerosi studi clinici. Ad esempio, uno studio pubblicato nel 1990 ha dimostrato che quando il Ketotifene e il Clenbuterolo vengono assunti insieme, si registra un significativo aumento della densità dei recettori beta-adrenergici rispetto all’uso del solo Clenbuterolo, che invece ne riduce di poco la densità in modo rapido. (2)Altri studi nei quali è stato usato il Salbutamolo hanno dimostrato che la riduzione dei recettori beta-adrenergici causata dall’utilizzo a lungo termine di questo agente potrebbe essere rapidamente invertita con appena 2 mg di Ketotifene fumarato al giorno. (3)
Se usato per ridurre la frequenza, la durata e la gravità degli attacchi d’asma, il Ketotifene fumarato viene solitamente somministrato inizialmente ad un dosaggio di 1 mg due volte al giorno (2 mg in totale). Se necessario, questo dosaggio può essere aumentato ad un dosaggio massimo di 2 mg due volte al giorno (4 mg totale). Gli atleti usano comunemente un dosaggio di 1 mg due volte al giorno (2 mg in totale) per l’uso (off-label) per la prevenzione della sottoregolazione dei beta-recettori dovuta all’uso di composti beta-agonisti come il Clenbuterolo e il Salbutamolo. Ciò può consentire a un individuo di ottenere un maggiore effetto termogenico, e di poter svolgere cicli più lunghi con farmaci beta-2 agonisti. Si noti che, data la capacità del Ketotifene di aumentare la sensibilità ai farmaci beta-agonisti, può essere necessario ridurre il dosaggio di questi ultimi se co-somministrati con Ketotifene fumarato.
L’emivita del Ketotifene fumarato è di 12 ore.
Gli effetti collaterali comuni con l’uso del Ketotifene fumarato includono secchezza delle fauci, stimolazione dell’appetito, aumento di peso, vertigini, stimolazione del SNC e sonnolenza. Questi effetti collaterali sono tutti comunemente associati a composti antistaminici forti. In rari casi possono verificarsi reazioni allergiche gravi a livello cutaneo o un’infiammazione vescicale chiamata cistite. Altri effetti collaterali connessi all’uso del Ketotifene fumarato comprendono Cefalea, nausea, vomito, dispepsia, e epigastralgia.
L’uso di Ketotifene fumarato deve essere evitato in soggetti sotto trattamento con antidiabetici orali e in caso di epilessia.
Il Ketotifene fumarato è ampiamente disponibile e viene venduto sotto numerosi nomi commerciali in molti paesi. La contraffazione su larga scala di questo medicinale non è attualmente nota da rappresentare un problema.
La pianta Artemisia dracunculus contiene sostanze che inibiscono la disgregazione muscolare nei diabetici obesi – e possono promuovere lo sviluppo muscolare, secondo studi sugli animali pubblicati dai ricercatori del Pennington Biomedical Research Center negli Stati Uniti. (1)
Artemisia dracunculus
L’Artemisia dracunculus è il Dragoncello, una comune erba usata in cucina. Gli atleti la conoscono come Dragoncello Russo. I produttori di supplementi talvolta combinano l’estratto di Artemisia dracunculus con la Creatina.
L’estratto usato dai ricercatori di Pennington per i loro esperimenti era il PMI 5011. Per evitare confusioni, diciamo subito che il PMI 5011 non è interessante per i produttori di integratori. L’equivalente umano delle dosi che i ricercatori hanno somministrato ai topi si aggirerebbe ad un quantitativo tra i 10 e 15g al giorno.
Gli ingredienti attivi nel PMI 5011 sono probabilmente i calconi. Se i ricercatori possono spiegare quali sono questi calconi e se sul mercato sono presenti integratori affidabili contenenti calconi , gli atleti potrebbero scoprire un nuovo integratore che vale la pena provare. Ed è di probabile interesse per gli atleti che usano Insulina esogena.
Pericolo
Se si sta pensando di fare da se gli estratti di Artemisia dracunculus, bisogna assicurarsi di farlo con le dovute precauzioni e conoscenze. L’Artemisia dracunculus contiene alcuni composti – come l’estragolo e il metyleugenolo – che sono pericolosi nella loro forma pura e in alte concentrazioni. Queste sostanze andranno quindi rimosse dal proprio estratto.
Muscoli
I diabetici che sono anche seriamente in sovrappeso possono sperimentare una accelerazione della perdita di massa muscolare in età avanzata. Il funzionamento del recettore dell’insulina si deteriora e i muscoli cominciano ad autodistruggersi. I processi molecolari coinvolti nella scomposizione delle proteine muscolari iniziano ad agire in maniera più marcata e più veloce.
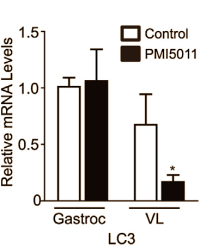
Heather Kirk-Ballard ha pubblicato uno studio su Nutrition in cui viene riportato che l’Artemisia dracunculus disattiva il gene per la catena leggera 3 della proteina 1 associata ai microtubuli [LC3] nel vasto laterale di topi diabetici.(2) La LC3 gioca un ruolo chiave nella disgregazione muscolare.
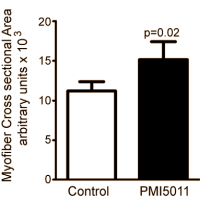
Quattro anni fa Kirk-Ballard ha pubblicato uno studio svolto su animali sul PLoS One nel quale veniva dimostrato che i topi con diabete avevano uno sviluppo muscolare maggiore quando il loro cibo conteneva l’1% di PMI 5011. (3)
Attraverso i numerosi studi sull’effetto anticatabolico di Artemisia dracunculus che Pennington ha realizzato negli ultimi anni, si sta prestando maggiore attenzione al PMI 5011. (4) (5) (6) (7) (8) (9) (10) (11)
Speculazioni
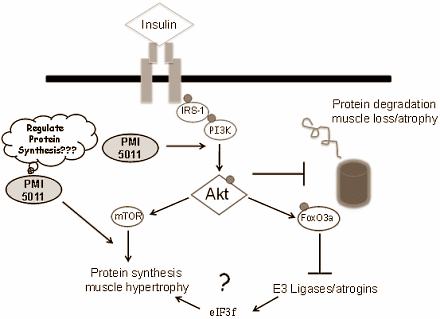
La figura seguente, che proviene dalla tesi di dottorato di Kirk-Ballard (12) pubblicata nel 2012, riassume come probabilmente funziona il PMI 5011.
Gli atleti utilizzatori di insulina esogena notano una perdita di efficacia del composto causata dal peggioramento dell’insulino-resistenza. Le loro cellule muscolari iniziano a perdere la loro sensibilità all’insulina, in un modo che assomiglia a ciò che accade ai tessuti muscolari nei diabetici obesi. Può darsi che gli ingredienti attivi dell’ Artemisia dracunculus possano rallentare questo processo. O lo impediscano.
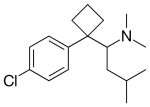
La Sibutramina (commercializzata come Meridia negli USA, Leptos in India, Reductil in Europa e altrove), solitamente in forma di idrocloruro monoidrato, è un riduttore dell’appetito per via orale, usato nel trattamento dell’obesità. È un inibitore del reuptake della serotonina e della noradrenalina ma non della dopamina; strutturalmente è simile alle amfetamine (1) anche se agisce diversamente. La Sibutramina è chimicamente una miscela racemica di (+) e (-) enantiomeri di 1- (4-clorofenil) -N, N-dimetil-α- (2-metilpropil)-cyclobutanemetanammina.
La Sibutramina esercita un effetto sulla perdita di peso attraverso due meccanismi distinti. Ha una marcata capacità di sopprimere l’appetito. Durante alcuni studi, i pazienti hanno ridotto la loro assunzione energetica di ben 1.300 Kcal durante l’assunzione di questo farmaco. (2) Oltre ai suoi effetti sull’assunzione calorica, la Sibutramina stimola anche il metabolismo e la spesa calorica giornaliera. Una singola dose di 10 mg ha dimostrata di aumentare il tasso metabolico basale fino al 30%, un effetto che viene mantenuto per almeno sei ore. Questa azione termogenica avviene attraverso l’interazione della molecola con il sistema adrenergico, soprattutto attraverso il supporto indiretto dell’attivazione dei recettori beta 3. Con l’uso di questo farmaco, si nota un forte aumento della termogenesi del tessuto adiposo marrone (BAT), che è accompagnato da aumenti della temperatura corporea di 0,5-1 grado Celsius.(3) L’elevazione della temperatura corporea è un buon indicatore dell’aumento della termogenesi.
Per avere un idea più chiara sull’efficacia della Sibutramina, si fa riferimento ad alcuni studi clinici svolti su questo agente. Uno di questi studi è stata condotta presso la Kansas Foundation for Clinical Pharmacology nel 2001. In questo studio, un gruppo di 322 pazienti obesi è stato trattato con 20 mg di Sibutramina o un placebo una volta al giorno per 24 settimane. Alla conclusione di questo studio, il 42% dei pazienti nel gruppo Sibutramina aveva perso il 5% o più del peso corporeo iniziale, mentre il 12% aveva osservato una perdita del peso corporeo del 10% o superiore. La Sibutramina è stata anche associata a significativi miglioramenti nei trigliceride e nei livelli di colesterolo HDL nel siero, in soggetti che presentavano valori scadenti all’inizio dello studio. Un altro studio dettagliato è stata svolto in Cina da parte del Dipartimento di Endocrinologia dell’ospedale Rui-jin nello stesso anno coinvolgendo un gruppo di 120 persone (uomini e donne) i quali sono stati trattati con soli 10 mg al giorno di Sibutramina. (4) Anche i risultati di questo studio sono stati favorevoli, con una media nella perdita di peso dei pazienti trattati pari a 15Kg nelle 24 settimane di utilizzo.
La Sibutramina ha ricevuto l’approvazione dalla Food and Drug Administration per la vendita come agente da prescrizione per la perdita di peso nel 1998. È stata sviluppata e commercializzata dalla Abbott Laboratories, che ha messo in vendita il farmaco sul mercato americano con il nome commerciale di Meridia. L’azienda ha anche venduto il farmaco in numerosi mercati internazionali sotto il nome di Reductil. La Sibutramina godette di un periodo limitato di vendite negli Stati Uniti, in quanto venne rimossa dal mercato nell’ottobre 2010 sotto pressione della FDA, citando una maggiore incidenza di eventi cardiovascolari avversi. La Abbott ritirò la Sibutramina da molti mercati in diverse aree del mondo. Si noti che la Sibutramina rimane classificata come sostanza controllata negli Stati Uniti. In Italia, nel marzo 2002, il Ministero della Salute ne decretò la sospensione dal commercio a seguito di una revisione dei dati di sicurezza, che rivelarono l’esistenza di un rischio cardiovascolare correlato al suo utilizzo. (5) Contemporaneamente l’Italia avviò una procedura di arbitrato a livello europeo richiedendo un parere al Comitato tecnico-scientifico dell’Agenzia europea per i medicinali. L’agenzia europea diede parere favorevole al mantenimento in commercio del farmaco, pertanto ad agosto dello stesso anno la sibutramina fu riammessa, con l’obbligo tuttavia di presentare ricetta medica specialistica per l’acquisto e di consegna al paziente di una scheda informativa sui possibili rischi.(6) L’agenzia europea aveva tuttavia richiesto all’azienda produttrice del farmaco uno studio multicentrico che valutasse l’efficacia e la sicurezza del farmaco. Lo studio richiesto venne denominato SCOUT (Sibutramine Cardiovascular OUTcome) e i risultati, pubblicati nel 2009, rivelarono che:
la perdita di peso ottenuta con la sibutramina è modesta
tale perdita di peso non si mantiene dopo la fine del trattamento
i pazienti trattati con Sibutramina avevano un aumento del rischio cardiovascolare del 16% rispetto ai pazienti trattati con placebo.
Il 24 gennaio 2010 pertanto, la Sibutramina venne nuovamente sospesa dalla commercializzazione.
La casa farmaceutica produttrice ha dichiarato di voler fare appello contro questa decisione.(7)
La Sibutramina cloridrato, dove ancora commercializzata, è comunemente venduta in capsule da 5 mg, 10 mg e 15 mg.
L’effetto collaterale più comune con la Sibutramina è l’aumento della pressione sanguigna, un tratto che lo rende un farmaco controindicato per l’utilizzo nei soggetti con pressione alta o altri problemi cardiovascolari. Altri effetti collaterali comuni sono la secchezza delle fauci, l’insonnia, l’irritabilità, il dolore alla schiena, il mal di stomaco e la stitichezza, che tendono a diminuire in termini di gravità quando l’utilizzatore si abitua al farmaco. La Sibutramina cloridrato deve essere sospesa immediatamente se si verificano effetti collaterali o sintomi più gravi di tossicità, inclusi eccitazione, irrequietezza, perdita di coscienza, confusione, agitazione, debolezza, brividi, fastidio, battito cardiaco accelerato, dilatazione delle pupille, vomito, difficoltà respiratorie, Dolori al petto, gonfiore di piedi, caviglie o gambe, svenimenti, disorientamento, depressione, febbre alta, dolore agli occhi, tremori o sudorazione eccessiva. Si ricordi che un’aumentata incidenza di eventi cardiovascolari avversi ha portato al ritiro di questo farmaco dalla maggior parte dei mercati. Non è generalmente più considerato un prodotto sicuro.
La Sibutramina cloridrato è usata per il trattamento dell’obesità, compresa la perdita e il mantenimento del peso, e deve essere utilizzata in combinazione con una dieta a ridotto apporto calorico. Questo farmaco è stato usato su pazienti con fattori di rischio aggiuntivi correlati al peso, tra cui ipertensione controllata, diabete e dislipidemia (colesterolo alto). Il dosaggio iniziale raccomandato per la maggior parte dei pazienti è 10 mg una volta al giorno, che deve essere regolato fino a 15 mg dopo 4 settimane se la perdita di peso non è stata inizialmente sufficiente. Di solito non sono raccomandate dosi più elevate. Anche gli atleti hanno usato questo farmaco nelle fasi di restrizione calorica, soprattutto per il suo effetto anoressizzante, ai medesimi dosaggi sopra indicati.
Ricordo inoltre che, questo farmaco è soggetto a disponibilità limitata a seguito di un richiamo sostenuto dalla FDA negli Stati Uniti e successivamente alla sua rimozione da molti altri mercati internazionali per ragioni di sicurezza.
Gabriel Bellizzi
Riferimenti:
–William Llewellyn’s ANABOLICS, 10th ed.
New Drugs, in Australian Prescriber, vol. 25, nº 1, 2002, p. 22. PDF
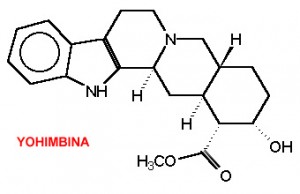
La Yohimbina (17α-hydroxy-yohimban-16α-carboxylic acid methyl ester) è un alcaloide estratto dalla corteccia della Pausinystalia Johimbe e dalle foglie della Rauvolfia serpentina (nota anche come radice di serpente indiana) venduto in alcuni paesi dietro prescrizione medica per il trattamento della disfunzione erettile, per il trattamento del disturbo da ridotto desiderio sessuale (riduzione della libido), e viene anche utilizzata per i suoi presunti effetti afrodisiaci.
La Yohimbina è un noto antagonista degli adrenocettori α-2. Ciò significa semplicemente che la Yohimbina può bloccare i recettori sulle cellule adipose che normalmente bloccano il processo di rilascio dei grassi nel torrente ematico. Ciò è controllato dagli ormoni Adrenalina e Noradrenalina (detti anche Epinefrina e Norepinefrina) che normalmente si legano agli adrenocettori delle cellule adipose per segnalare il rilascio dei grassi nel sistema circolatorio affinché possano essere bruciati come fonte energetica. Il corpo cerca di fermare questo processo attraverso la stimolazione dei recettori α-2 che dicono alle cellule grasse di accumulare materiale. Quando la Yohimbina blocca i recettori α-2 viene rilasciato più grasso che poi viene bruciato come fonte energetica.
Il principale studio che supporta quanto sopraddetto è “Does yohimbine act as a slimming drug?” di Kucio C., Jonderko, K., Piskorska D., Dipartimento di gastroenterologia, Silesian School of Medicine, Katowice, Polonia [Isr J Med Sci. 1991 Oct;27(10):550-6].
Dal momento che la Yohimbina è un antagonista degli α2-recettori, è stata esaminata per la sua efficacia nella cura dell’obesità.
20 donne obese sono state sottoposte a 3 settimane di dieta ipocalorica (1.000 kcal/giorno) dopo di che sono state assegnate a caso a due trattamenti secondo la procedura dello studio a doppio cieco: 10 soggetti hanno ricevuto 5mg di Yohimbina 4 volte al giorno mentre altri 10 hanno ricevuto un placebo, tutti hanno seguito una dieta ipocalorica di 1.000 kcal/giorno.
I risultati di questo studio hanno mostrato che i soggetti che hanno ricevuto la Yohimbina hanno sperimentato una perdita di grasso media di 3,55 kg mentre il gruppo placebo ha perso solo 2,21 kg. Questi studi hanno mostrato anche che il rilascio di Insulina dovuto all’ingestione del cibo inibisce le azioni della Yohimbina mentre una dieta molto ipocalorica ne agevola l’efficacia. Meno cibo significa anche meno rilascio di Insulina. Quindi una dieta ipocalorica combinata con una somministrazione multidose di Yohimbina può aumentare il dispendio calorico di oltre il 30%.
Ci sono almeno due punti chiave per massimizzare gli effetti della Yohimbina:
1. Una dose adeguata– Molte volte viene consigliata una dose troppo bassa. Negli studi, la quantità minima di Yohimbina assunta oralmente è stata di 0,2 mg/kg al giorno. Quindi una persona di 90 kg ha bisogno di 18 mg di Yohimbina al giorno per ottenere l’effetto lipolitico desiderato. Attenzione: con la Yohimbina alcune persone sperimentano delle risposte molto forti del ritmo cardiaco e della pressione ematica. Per valutare la propria tolleranza è consigliabile cominciate con circa 1/3 della dose ottimale (quindi una persona di 90 kg comincerebbe con 6 mg) incrementando la dose nel corso di un periodo di alcuni giorni (generalmente 2-3 giorni). Se il ritmo cardiaco o la pressione ematica aumentano troppo si consiglia l’interruzione dell’assunzione di Yohimbina.
2. Deve essere assunta a stomaco vuoto– È una cosa fondamentale perché anche una piccola risposta insulinica impedirà qualsiasi effetto della Yohimbina. Assumere la Yohimbina con il cibo produrrà una risposta insulinica maggiore rispetto all’assunzione del cibo da solo (la cosa è legata ai recettori α nel pancreas). In base a questa regola, uno dei momenti migliori per assumere la Yohimbina è prima dell’allenamento aerobico del mattino. Se non si svolge una sessione aerobica al mattino, credo che un altro momento favorevole per assumere la Yohimbina sia 30 minuti prima della sessione di allenamento serale, o comunque almeno 3-4 ore dopo aver mangiato affinché i livelli di Insulina calino a sufficienza. Comunque penso che la prima parte della giornata sia il momento migliore.
Alcuni atleti assumono la Yohimbina con un po’ di Caffeina circa 30 minuti prima della sessione aerobica. Assumono anche l’associazione ECA, ma mai insieme alla Yohimbina. La combinazione ECA + Yohimbina, come l’associazione Yohimbina + Clenbuterolo, è potenzialmente pericolosa (più dei due composti presi singolarmente) a causa dell’interazione sul ritmo cardiaco e sulla pressione ematica. Gli atleti in questione assumono la Yohimbina prima della sessione aerobica del mattino, e per la prima dose di ECA aspettano 4 ore dopo l’assunzione di Yohimbina. L’emivita della Yohimbina è di 0,25-2 ore.
Ricapitolando, gli effetti collaterali della Yohimbina a seconda del dosaggio assunto possono comportare anche importanti alterazioni della pressione arteriosa, in senso ipertensivo, che spesso si accompagnano ad aumento della frequenza cardiaca. Gli eventi avversi a carico del tratto gastrointestinale comprendono nausea e vomito. Per i suoi effetti eccitatori a carico del SNC la molecola comporta spesso la comparsa di irritabilità, ansia generalizzata, eccitazione, iperattività motoria, reazioni maniacali, allucinazioni, insonnia, tremore e vertigine. La contemporanea assunzione di Yohimbina e farmaci antidepressivi o fenotiazine può comportare un aumentato rischio di comparsa e aggravamento degli effetti collaterali.
Concludendo, la Yohimbina si presenta come un efficace agente lipolitico specie nei confronti del grasso testardo. Dati i suoi possibili effetti collaterali, la Yohimbina deve essere usata con cautela calcolando accuratamente le dosi così da evitare o diminuire la comparsa di effetti avversi.
Gabriel Bellizzi
Riferimenti
– Domande & Risposte senza censura di Author L. Rea (Olympian’s News)
– Chemical Muscle Enhancement II (Author L. Rea)
– Chiedete a Lyle McDonald (THINK MUSCLE – Olympian’s News)
Gli AAS (in base oleosa e acquosa) possono essere iniettati sottocute, anche se la loro somministrazione principalmente consigliata è tramite iniezione intramuscolare. Le iniezioni di AAS che utilizzano il tessuto adiposo sottocutaneo come luogo di deposito del farmaco modificano soltanto il tasso di rilascio dello steroide dal sito di iniezione, anche se non è stata dimostrata molta differenza dalle iniezioni intramuscolari. Gli individui che …vogliono assumere un AAS tramite iniezione sottocutanea dovrebbero fare attenzione al quantitativo della soluzione, poiché il tessuto sottocutaneo non può contenere un volume di olio iniettato troppo alto senza creare problemi, arrivando a “tollerare” quantitativi decisamente inferiori rispetto alle iniezioni intramuscolari. Per quanto riguarda le affermazioni circa gli ascessi dovuti a tele metodica di iniezione, c’è da specificare che essi si verificano solo se il sito di iniezione è infetto o se il composto è contaminato.
Come risaputo, quando si parla di iniezioni sottocutanee si sta parlando di un metodo di iniezione che prevede la somministrazione di una soluzione (principalmente) a base acquosa nel tessuto adiposo appena sotto la pelle. Anche le soluzioni a base oleosa possono essere iniettate sottocute, ma non è normalmente raccomandato. Le iniezioni sottocutanee sono utilizzate soprattutto per la somministrazione di Insulina, GH (Ormone della Crescita), HCG (Gonadotropina Corionica Umana) e altri peptidi. Le iniezioni sottocutanee sono utilizzate principalmente per iniettare sostanze a base acquosa e composti che richiedono quantità della soluzione molto piccole (1mL o CC o meno), in quanto i siti di iniezione sottocutanei non possono contenere una quantità elevate di soluzione senza problemi. Gli AAS in base acquosa e oleosa possono quindi essere iniettati sottocute, ma prestando attenzione al volume dell’iniezione, iniettando un quantitativo molto inferiore rispetto alle iniezioni intramuscolari. Gli studi hanno mostrato livelli plasmatici stabili in seguito a somministrazione sottocutanea di AAS con efficienza tale ad una somministrazione intramuscolare. (1) (2)
Gabriel Bellizzi
Riferimenti:
1- Subcutaneous administration of testosterone. A pilot study report. Al-Futaisi AM, Al-Zakwani IS, Almahrezi AM, Morris D. Saudi Med J 2006;27(12):1843-6.
2- STABLE TESTOSTERONE LEVELS ACHIEVED WITH SUBCUTANEOUS TESTOSTERONE INJECTIONS. M.B. Greenspan, C.M. Chang. Division of Urology, Department of Surgery, McMaster University, Hamilton, ON, Canada.