Androgenico: 24
Anabolico: 322-630
Standard: Methyltestosterone (orale)
Nome Chimico: 17b-hydroxy-17a-methyl-2-oxa-5a-androstane-3-one
Attività estrogenica: nessuna
Attività Progestinica: nessuna
Aromatizzazione: no
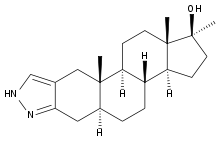
L’Oxandrolone ( 17β-idrossi-17α-metil-2-ossa-5α-androstan-3-one) è uno steroide di sintesi, derivato del Diidrotestosterone, con attività androgena pari a 24 ed anabolizzante pari a 630 rispetto al Testosterone (100/100). Differisce dal DHT per la metilazione in C-17 e per la sostituzione in C-2 di una molecola di ossigeno.
L’Oxandrolone è stato descritto per la prima nel 1962.(1) È stato sviluppato come farmaco diversi anni dopo dal gigante farmaceutico G.D. Searle & Co. (ora Pfizer), che l’ha commercializzato negli Stati Uniti e nei Paesi Bassi con il nome commerciale di Anavar. La Searle lo commercializzo (attraverso licenza) anche con nomi commerciali diversi, tra cui Lonavar (Argentina, Australia), Lipidex (Brasile), Antitriol (Spagna), Anatrophill (Francia), e Protivar. L’Oxandrolone è stato ideato per essere un anabolizzante orale estremamente mite, che potesse anche essere utilizzato in modo sicuro da donne e bambini. A questo proposito Searle ebbe successo, in quanto l’Anavar mostrò un alto grado di successo terapeutico e tollerabilità negli uomini, nelle donne e nei bambini. Durante i suoi primi anni, Anavar venne proposto per un certo numero di applicazioni terapeutiche, tra cui la promozione della crescita della massa magra durante malattie cataboliche, la promozione della crescita della massa magra dopo operazioni chirurgiche, traumi, infezioni, o somministrazione prolungata di corticosteroidi, o il supporto della densità ossea in pazienti con osteoporosi.
Nel 1980, l’FDA affinò le applicazioni approvate per l’Anavar includendo la promozione dell’aumento del peso dopo intervento chirurgico, infezione cronica, traumi, o perdita di peso senza motivo fisiopatologico definito. Nonostante la sua esperienza continua sulla sicurezza, Searle decise di interrompere volontariamente la vendita dell’Anavar il 1 ° luglio 1989. L’arresto delle vendite e la crescente preoccupazione dell’opinione pubblica circa l’uso degli steroidi anabolizzanti da parte degli atleti sembravano essere alla base di questa decisione. Con il marchio Anavar fuori dal mercato, l’Oxandrolone era completamente scomparso dalle farmacie degli Stati Uniti. Poco dopo, i prodotti conteneti Oxandrolone nei mercati internazionali (spesso venduti da o su licenza della Searle) hanno cominciato a scomparire, così, come il leader mondiale nella produzione del farmaco ha proseguito il suo ritiro dal commercio degli steroidi anabolizzanti. Per diversi anni, durante i primi anni 90, sembrava che l’Anavar potesse uscire definitivamente dal mercato.
Questo accadeva circa sei anni prima che le compresse di Oxandrolone tornassero sul mercato statunitense. Il prodotto venne reintrodotto sugli scaffali delle farmacie nel dicembre 1995, questa volta con il nome di Oxandrin della Bio-Technology Corp. (BTG). La BTG avrebbe continuato a venderlo con l’autorizzazione della FDA la quale lo aveva approvato per utilizzi che implicano la conservazione della massa magra, ma era stato anche concesso lo status di farmaco-orfano per il trattamento del deperimento derivante dall’AIDS, epatite alcolica, la sindrome di Turner nelle ragazze, e il ritardo costituzionale della crescita e della pubertà nei ragazzi. lo status di farmaco-orfano conferì alla BTG un monopolio di 7 anni sul farmaco per questi nuovi usi, permettendo ad essa di proporre un prezzo di vendita molto alto. Molti pazienti furono indignati di apprendere che il farmaco li sarebbe costato (al prezzo all’ingrosso) tra i 3.75 e i 30$ al giorno, che risultava molte volte più costosa rispetto all’Anavar solo pochi anni prima. Il rilascio di un compresse da 10 mg da parte della BTG diversi anni dopo fece ridurre il costo relativo del farmaco.
L’Oxandrin® continua ad essere venduto negli Stati Uniti, ma ora è sotto l’etichetta Savient (precedentemente noto come BTG). Esso è attualmente approvato dalla FDA per “terapia aggiuntiva per promuovere l’aumento di peso dopo perdita di peso in seguito ad grossi interventi chirurgici, infezioni croniche, o gravi traumi e, in alcuni pazienti che, senza precise ragioni fisiopatologiche non riescono a ottenere o mantenere un peso normale, per compensare il catabolismo proteico associato a somministrazione prolungata di corticosteroidi, e per il sollievo del dolore osseo spesso accompagnato dall’osteoporosi.” Versioni generiche del farmaco sono ora disponibili negli Stati Uniti, cosa che ha ridotto il prezzo della terapia con Oxandrolone. Al di fuori degli Stati Uniti, l’Oxandrolone rimane disponibile, anche se non ampiamente.
Come accennato in precedenza, l’Oxandrolone è una forma modificata del Diidrotestosterone. Differisce da questo per:
1) l’aggiunta di un gruppo metilico al carbonio in posizione 17-alfa per proteggere l’ormone durante il passaggio epatico in seguito a somministrazione orale;
2) la sostituzione con una molecola di ossigeno in C-2; l’Oxandrolone è l’unico steroide con tale sostituzione alla sua struttura nell’anello di base. Grazie a questa modifica ne derivano alcuni vantaggi:
– Notevole aumento della resistenza all’ossidazione
– Immunità della molecola alla deidrogenasi (precisamente alla 3-alfa-idrossi-deidrogenasi), che è la responsabile della pressoché istantanea deattivazione del Diidrotestosterone a livello recettoriale muscolare
– Diminuzione pressoché totale dell’affinità con le SHBG.
Queste caratteristiche aumentano considerevolmente la potenza anabolizzante della molecola.
L’Oxandrolone possiede una proprietà comune a diversi AAS orali, cioè la proprietà di abbassare le SHBG circolanti. Ciò può portare a due effetti:
1- Aumento del livello degli AAS liberi in circolo, specie quelli con una forte affinità per le SHBG (Testosterone, Drostanolone, Metenolone, ecc): la diminuzione è pari al 40% con Oxandrolone, 60% con Stanozololo e 70% con Chlorodehydromethyltestosterone.
2- Aumento dei livelli degli Estrogeni liberi circolanti con un aumentato rischio di sviluppare effetti estrogenici avversi(2)
Il punto “2” deve essere comunque preso in considerazione, sia quando l’Oxandrolone (o altra molecola avente la medesima caratteristica) viene assunto per un periodo superiore alle 4 settimane con molecole non aromatizzabili sia, a maggior ragione, quando assunto con molecole aromatizzabili.
L’Oxandrolone possiede un’altra caratteristica la quale comporta un impatto complessivo considerevole, la riduzione dell’attività del Cortisolo. Essa è dovuta all’agonismo inverso esercitato a livello dei recettori glucocorticoidei . Questo comporta, in seguito alla somministrazione di Oxandrolone, un aumento dei livelli di Cortisolo libero e legato, molto probabilmente proprio a causa della sua azione antagonista del recettore del Cortisolo (3).
Non vi sono prove in letteratura che indicano un interazione dell’Oxandrolone con il recettore del Progesterone.
L’Oxandrole è spesso inserito in fase di “Cut” o nel “Pre-Gara” dal momento che questa molecola fornisce considerevole sostegno in queste delicate fasi. Oltre all’inibizione dell’attività recettoriale corticosteroidea, la quale presenta un’azione preventiva sul catabolismo muscolare, l’Oxandrolone presenta:
– Una spiccata riduzione della prealbunina legante la Tiroxina (TBPA), con conseguente rialzo del T-3 circolante; ciò comporta un azione marcata del T-3 anche se questo è somministrato a dosi fisiologiche standard (25mcg) durante una dieta a forte componente ipocalorica-ipoglucidica(3).
– Aumento della chetogenesi epatica e conseguente stimolo della lipolisi.
Esiste uno studio del 2008 (4) nel quale si è osservato l’aumentò della Chetogenesi epatica in maschi adulti in seguito alla somministrazione di Oxandrolone: ai diciotto uomini che hanno partecipato allo studio è stata somministrata una dose di 10mg/die di Oxandrolone per più di una settimana. I soggetti avevano la valutazione della chetogenesi epatica all’inizio dello studio e dopo 7 giorni di somministrazione di Oxandrolone. La chetogenesi è stata valutata misurando i livelli plasmatici di 3-idrossibutirrato nel corso di un test di tolleranza lipidica. L’Oxandrolone portò ad un aumento dei livelli di 3-idrossibutirrato a digiuno del 70%, aumentando l’area sotto la curva nel corso di un FFT del 53% sopra i livelli di pretrattamento senza compromettere le aree sotto la curva per gli acidi grassi non esterificati, il glicerolo ed i trigliceridi. A digiuno i livelli di 3-idrossibutirrato erano correlati con gli acidi grassi non esterificati ed i trigliceridi. Lo studio ha dimostrato come già la somministrazione a breve termine di Oxandrolone dia come risultato un marcato aumento della chetogenesi epatica. Questo risultato è coerente con il maggior afflusso di acidi grassi nel fegato secondario alla lipolisi delle lipoproteine e all’incremento della lipasi epatica. Tuttavia, non si può escludere la possibilità che l’Oxandrolone agisca direttamente nel fegato per stimolare l’ossidazione degli acidi grassi.
Grazie a queste caratteristiche, l’Oxandrolone trova la sua più grande utilità se inserito durante una fase di “Cut” o “Pre-Gara”, dove il ridotto apporto calorico e (spesso) glucidico rendono queste fasi molto delicate. Un abbinamento in questo frangente può essere (per un atleta intermedio/avanzato) composto da Oxandrolone, Trenbolone e GW1516.
Esistono ulteriori prove dell’efficacia dell’Oxandrolone derivanti da uno studio nel quale si è osservata una riduzione di 4 Libre di massa grassa (2Kg circa), con un guadagno di 7 Libre di massa magra (3,5Kg circa) nel corso di 12 settimane di trattamento con soli 20 mg di Oxandrolone (10 mg, due volte al giorno) (5).
Inizialmente l’Oxandrolone veniva considerato uno steroide anabolizzante di Classe I, cioè con affinità recettoriale prevalentemente AR, essendo un DHT derivato, notoriamente marcatamente AR. Recentemente è stato scoperto che la sua affinità AR e molto bassa. Questa caratteristica è probabilmente dovuta, oltre alla metilazione in C-17 che aumenta l’affinità recettoriale non genomica, al fatto che l’Oxandrolone presenta una modifica dello scheletro del nucleo steranico (unica nella grande famiglia degli AAS commercializzati). La modifica in C-2 , con l’inserimento di una molecola di ossigeno, è criptata in un anello tipo A, e ciò lo fa spesso classificare come un AAS eterociclico, invece che un derivato del DHT.
Essendo l’Oxandrolone un DHT derivato esso non aromatizza in estrogeno, e non possiede attività estrogenica misurabile. L’Oxandrolone non presenta attività progestinica correlata.(6) Quindi, effetti collaterali quali ritenzione idrica e ginecomastia non sono generalmente un problema con l’uso di questa molecola.
Anche se classificato come uno steroide anabolizzante con un valore androgeno basso, gli effetti collaterali androgeni sono ancora possibili con questa molecola. Questo può includere acne, pelle oleosa e alopecia (dipendente dalle caratteristiche individuali). Per quanto riguarda l’uso di questa molecola in ambito femminile gli effetti androgeni possibili possono includere voce profonda, irregolarità mestruale, crescita di peli sul corpo e sul viso, cambiamenti nella struttura della pelle la crescita di peli sul viso, e l’allargamento del clitoride.
Essendo l’Oxandrolone uno steroide con bassa attività androgenica rispetto alle sue azioni anabolizzanti, la soglia dei possibili forti effetti collaterali androgeni è relativamente superiore se confrontato con agenti più androgeni come il Testosterone, Methandrostenolone o Fluoxymesterone.
Come già accennato, l’Oxandrolone è un composto metilato in posizione C-17. Questa alterazione protegge la molecola dalla disattivazione epatica, permettendo ad una percentuale molto elevata del farmaco di entrare nel sangue dopo somministrazione orale. Gli AAS C17-alfa alchilati esercitano azione epatotossica. L’esposizione prolungata o ad alte dosi può causare danni al fegato. In rari casi si viene a creare una pericolosa disfunzione epatica. Si consiglia perciò un controllo regolare durante ogni ciclo monitorando la funzionalità epatica e la salute generale. L’assunzione di AAS c17-alfa alchilati non dovrebbe mai superare le 6-8 settimane di assunzione continua, nel tentativo di evitare un eccessivo stress epatico.
Nonostante l’Oxandrolone sia un AAS metilato in C-17 sembra causare un minore stress epatico rispetto ad altre molecole c-17 alfa alchilate. Il produttore del farmaco descrive la molecola di Oxandrolone come uno steroide che non viene ampiamente metabolizzato dal fegato come accade per le altre forme di AAS orali metilati in C-17 sperimentati, e ciò può essere un fattore determinante nella sua ridotta epatotossicità. Ciò è dimostrato dal fatto che più di un terzo del composto è ancora intatto quando escreto nelle urine.(7)
In uno studio si è voluto confrontare gli effetti dell’Oxandrolone con altri agenti C-17 alfa alchilati tra cui Methyltestosterone, Noretandrolone, Fluoxymesterone e Methandriol dimostrando che l’Oxandrolone provoca una ritenzione di sulfobromofthaleina più bassa (BSP, un marker dello stress epatico) rispetto agli agenti testati.(8) 20 mg di Oxandrolone hanno prodotto il 72% in meno di ritenzione del BSP rispetto ad un dosaggio uguale di Fluoxymesterone, e ciò è una notevole differenza essendo entrambe molecole 17-alfa alchilate.
Uno studio più recente ha esaminato la risposta a dosi crescenti (20 mg, 40 mg e 80 mg) di Oxandrolone in 262 pazienti maschi affetti da HIV. Il farmaco è stato somministrato per un periodo di 12 settimane. Il gruppo che assumeva 20 mg di Oxandrolone al giorno non ha mostrato tendenze statisticamente significative di epatotossicità esaminando i valori degli enzimi epatici (AST / ALT; amino-transferasi e alanina amino-transferasi). Gli uomini che assumevano 40 mg hanno sperimentato un aumento medio di circa il 30-50% dei valori degli enzimi epatici, mentre il gruppo degli uomini che assumevano 80 mg hanno sperimentato un aumento di circa il 50-100%. Circa il 10-11% dei pazienti del gruppo che assumeva 40 mg di Oxandrolone hanno sperimentato un livello di tossicità epatica del III e IV grado della tossicità in base ai valori AST e ALT secondo le linee dell’Organizzazione Mondiale della Sanità . Questa cifra è passata al 15% nel gruppo che assumeva 80 mg di Oxandrolone. Mentre l’epatotossicità grave non può essere esclusa con Oxandrolone, questi studi suggeriscono che questa molecola fornisce un margine di sicurezza maggiore rispetto ad altri agenti metilati in C-17.(9) L’uso di un adeguata epatoprotezione durante l’uso di questa molecola è comunque consigliata.
Come risaputo, gli AAS possono avere effetti deleteri sul colesterolo sierico. Questo include una tendenza alla riduzione delle concentrazioni di colesterolo HDL (buono) e un aumento delle concentrazioni di colesterolo LDL (cattivo), cosa che comporta uno sbilanciamento dell’equilibrio HDL/LDL che si traduce in un rischio maggiore di sviluppare arteriosclerosi. L’impatto relativo all’assunzione di un AAS nei confronti dei lipidi ematici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizable o non aromatizable), e dal livello di resistenza al metabolismo epatico. L’Oxandrolone ha un forte effetto sulla gestione epatica del colesterolo a causa della sua resistenza strutturale durante il passaggio epatico, dalla sua natura non aromatizable, e per la via di somministrazione. Nello studio precedentemente citato svolto su soggetti di sesso maschile malati di HIV, 20 mg di Oxandrolone al giorno per 12 settimane hanno causato una riduzione media del 30% dei livelli serici di HDL. I valori di HDL sono stati soppressi del 33% nel gruppo a 40 mg e del 50% nel gruppo a 80 mg. Questa riduzione è stata accompagnata da un aumento statisticamente significativo nei valori di LDL (circa 30-33%) nel gruppo a 40 mg e in quello a 80 mg, aumentando ulteriormente il rischio aterogenico. Gli AAS possono influenzare negativamente la pressione sanguinea, le concentrazioni di trigliceridi, ridurre il rilassamento endoteliale, e promuovere l’ipertrofia ventricolare sinistra, tutti potenziali fattori che aumentano il rischio di sviluppare malattie cardiovascolari e infarto del miocardio.
Inizialmente si pensò che l’Oxandrolone potesse divenire un possibile farmaco per chi soffre di squilibri di colesterolo e/o trigliceridi. I primi studi hanno mostrato che la molecola era in grado di abbassare i valori di colesterolo totale e dei trigliceridi in alcuni tipi di pazienti iperlipidemici, facendo pensare ai ricercatori che il farmaco potesse avere un potenziale come agente ipolipemizzante.(10) In seguito ad ulteriori indagini è stato accertato, tuttavia, che un abbassamento dei valori di colesterolo totale in seguito a somministrazione di Oxandrolone era accompagnato da una ridistribuzione nel rapporto del colesterolo buono (HDL) a favore del colesterolo cattivo (LDL) cosa che favorisce un maggiore rischio aterogenico.(11,12)
Questo nega alcun effetto positivo che questo farmaco possa avere sui trigliceridi o il colesterolo totale, e in realtà lo rende potenzialmente pericoloso in termini di rischio cardiaco, soprattutto se assunto per periodi di tempo prolungati. Oggi ci rendiamo conto di come gli AAS possano produrre variazioni sfavorevoli del profilo lipidico, e non sono molto utili nei disordini del metabolismo lipidico. Ciò che è certo è che essendo l’Oxandrolone un AAS metilato in c17 assunto oralmente, è ancora più rischioso da usare a questo proposito di un iniettabile esterificato, come un Testosterone o Nandrolone.
Per contribuire a ridurre lo sforzo cardiovascolare si consiglia di mantenere un programma di esercizio cardiovascolare e ridurre al minimo l’assunzione di grassi saturi, colesterolo e carboidrati semplici in ogni momento durante la somministrazione di AAS. Supplementando con oli di pesce (4 grammi al giorno) e un integratore naturale per il controllo del colesterolo come la Niacina.
Tutti gli AAS se assunti in dosi sufficienti per promuovere la crescita muscolare causano una soppressione della produzione di Testosterone endogeno. L’Oxandrolone non fa eccezione. Nello studio sopra citato svolto su soggetti di sesso maschile malati di HIV, la somministrazione per dodici settimane di 20 mg o 40 mg al giorno di Oxandrolone ha causato una riduzione di circa il 45% dei livelli sierici di Testosterone. Il gruppo che assumeva 80 mg ha subito una riduzione del 66% del Testosterone. Tendenze simili di soppressione sono state notate nella produzione di LH, con le dosi di 20 mg e 40 mg causando una riduzione del 25-30%, mentre nel gruppo 80 mg è stato notato un calo di oltre il 50%. Inoltre, gli studi sui ragazzi con ritardo della pubertà costituzionale hanno mostrato una significativa soppressione di LH e Testosterone endogeno con una dose minima di 2,5 mg al giorno.(13)
In effetti, uno studio di Sheffield-Moore et al. ha dimostrato che una dose di soli 15mg/die di Oxandrolone in giovani uomini sani per cinque giorni ha ridotto i livelli di testosterone totale e libero(14).
Senza lo svolgimento di una adeguata PCT, i livelli di Testosterone dovrebbe tornare alla normalità entro 1-4 mesi dalla cessione dell’assunzione del AAS. Si noti che un ipogonadismo ipogonadotropo può sviluppare secondariamente all’abuso di steroidi, cosa che richiede un intervento medico.
L’Oxandrolone è disponibile solo in alcuni mercati come farmaco per uso umano. la composizione e il dosaggio può variare a seconda del paese e del produttore. Il marchio originale Anavar conteneva compresse da 2,5 mg di principio attivo. L’Oxandrin contiene compresse da 2,5 mg o 10 mg di principio attivo. Altri marchi attuali contengono comunemente 2,5 mg, 5 mg o 10 mg di principio attivo per compressa. Sul mercato nero delle UGL è largamente commercializzato, anche se con dubbia legittimità del prodotto.
La questione del dosaggio è come sempre soggettiva, calcolabile tenendo presente:
1- I dosaggi complessivi degli altri AAS (eventualmente) co-somministrati e;
2- che l’effetto anabolizzante di questa molecola cresce logaritmicamente in modo pressoché lineare fino a 0,75mg/Kg aumentando sensibilmente fino ad 1mg/Kg per poi arrestarsi a dosaggi superiori, e che una dose di 0,55mg/Kg risulta più funzionale e gestibile (sia a livello di effetti avversi che guadagni complessivi), specie se la molecola è co-somministrata con altri AAS.(15)
Per le donne i dosaggi maggiormente funzionali si sono attestati tra i 5-10mg ed i 15-20mg al giorno.
E’ utile ricordare che studi hanno dimostrato che l’assunzione di uno steroide anabolizzante per via orale con il cibo può diminuire la sua biodisponibilità.(16) Ciò è causato dalla natura liposolubile degli ormoni steroidei, che permette ad una parte del farmaco di sciogliersi con i grassi alimentari non digeriti, riducendo il suo assorbimento dal tratto gastrointestinale. Per il massimo assorbimento, anche questo AAS dovrebbe essere assunto a stomaco vuoto.
L’Oxandrolone può essere identificato positivamente mediante il test di sostanze ROIDTEST ™ B & C. Dopo le recenti tendenze del mercato, troviamo che i preparativi del mercato nero etichettatati come “Oxandrolone ” hanno un alto rischio di non contenente lo stesso o di contenere altri steroidi. La probabilità che ciò avvenga è particolarmente alta con questo AAS visti gli alti costi delle polveri.
Il kit ROIDTEST ™ può essere utilizzato per confermare la presenza di questo AAS in un prodotto, e può essere acquistato qui:

Gabriel Bellizzi
Note:
1) Oxandrolone: A Potent Anabolic Steroid of Novel Chemical Composition. Fox M, Minot AS, and Liddle GW. Journal of Clinical Endocrinology and Metabolism. 1962; Volume 22, Pgs. 921-924.
2) Wasserman P, Segal-Maurer S, Rubin D. Low sex hormone-binding globulin and testosterone levels in association with erectile dysfunction among human immunodeficiency virus-infected men receiving testosterone and oxandrolone. J Sex Med, 2008;5(1):241-7.
3) Barbosa J, Seal US, Doe RP: Effects of anabolic steroids on hormone-binding proteins, serum cortisol and serum nonprotein-bound cortisol. J Clin Endocrinol Metab, 1971;32(2):232-40.
4) Vega GL, Clarenbach JJ, Dunn F, Grundy SM. Oxandrolone enhances hepatic ketogenesis in adult men. J Investig Med, 2008;56(7):920-4.
5) Schroeder ET, Zheng L, Ong MD, Martinez C, Flores C, Stewart Y, Azen C, Sattler FR: Effects of androgen therapy on adipose tissue and metabolism in older men. J Clin Endocrinol Metab, 2004;89(10):4863-72.
6) Published reference of personal communication from Saunders F.J. (April 21, 1961) to author of Methyltestosterone, related steroids, and liver function. Arch Int. Med 116 (1965):289-94.
9) William Llewellyn’s ANABOLICS, 10th ed.
10) Effects of Oxandrolone on Plasma Lipoproteins and the Intravenous Fat tolerance in Man. Atherosclerosis 19 (1974):337-46.
12) Plasma and Lipoprotein Lipid Responses to Four Hypolipid Drugs. Lipids 19 (1984):73-79.
13) The effects of oxandrolone on the growth hormone and gonadal axis in boys with constitutional delay of growth and puberty. Malhitra A, Poon E. Et al. Clin Endocrinol (Oxf ) 1993 Apr;38(4):393-8.
14)Sheffield-Moore M, Urban RJ, Wolf SE, Jiang J, Catlin DH, Herndon DN, Wolfe RR, Ferrando AA: Short-term oxandrolone administration stimulates net muscle protein synthesis in young men. J Clin Endocrinol Metab, 1999;84(8):2705-11.
15) Costruire la bestia perfetta: Chemical muscle enhancement 2. di
16) Anabolic Steroids and Sports Volume II. James E. Wright. Sports Science Consultants, Natick, MA 1982.