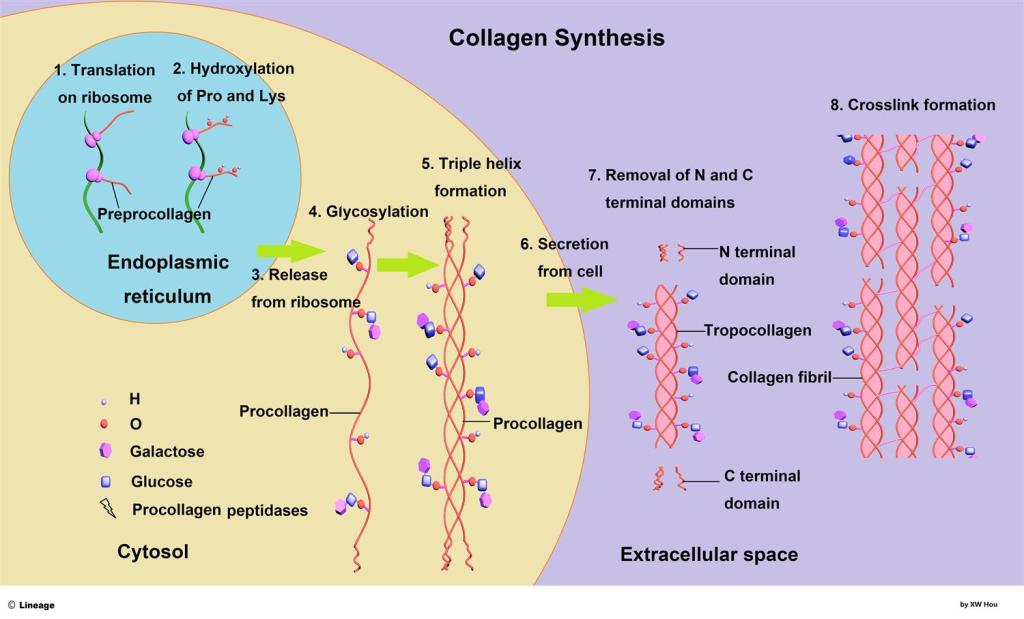
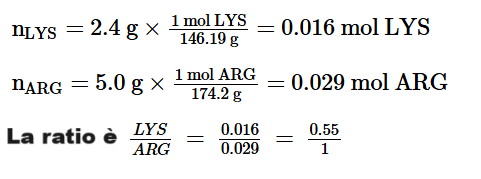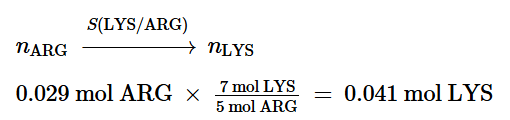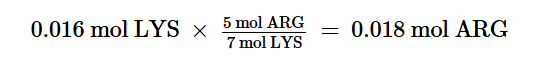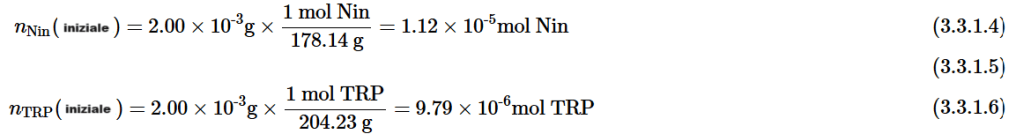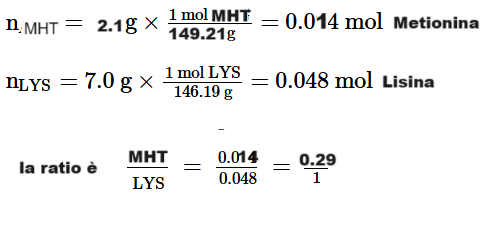Nella prima parte di questa disamina sono state passate in rassegna diverse informazioni necessarie per comprendere cosa sono le Incretine e quali sono le loro principali azioni. Si è poi passati a descrivere la classe di farmaci degli Incretino-Mimetici discorrendo sulla loro sintesi, tipologia molecolare [in ordine cronologico in base alla data di commercializzazione] e caratteristiche di azione sia per quanto concerne le attività positive che quelle negative. In conclusione, si è accennato alla consistente diffusione dell’uso “cosmetico” di tali farmaci.
In questa seconda ed ultima parte vedremo come questa relativamente nuova classe di farmaci abbia trovato un certo spazio di diffusione nel Fitness e (sebbene in minor parte) nel BodyBuilding. Verranno discussi i punti di attrattiva e le limitazioni (e rischi) legati al loro uso.
Le “attrattive” degli Incretino-Mimetici:
L’obesità è una grave epidemia che affligge la società del così detto “occidente americanizzato”. I farmaci iniettabili sottocutanei inizialmente concepiti per la gestione del diabete di tipo II, come la Semaglutide e altri agonisti del recettore del GLP-1, stanno rapidamente guadagnando popolarità per i loro effetti sulla perdita di peso. Questi farmaci (Ozempic, Wegovy, Saxenda e Mounjaro) sono onnipresenti sui social media e sono promossi da celebrità di tutte le fasce demografiche. “Viso da Ozempic” e ‘sedere da Ozempic’ sono ormai concetti mainstream che evidenziano i possibili cambiamenti morfologici che si verificano con questi farmaci. Con la diffusione non controllata da personale qualificato dell’uso di questi farmaci, è aumentato anche l’elenco dei potenziali effetti avversi.
Nella prima parte di questa disamina si è constatato che gli Incretino-Mimetici hanno potenzialità che si esprimono su modifiche della composizione corporea e che riguardano principalmente:
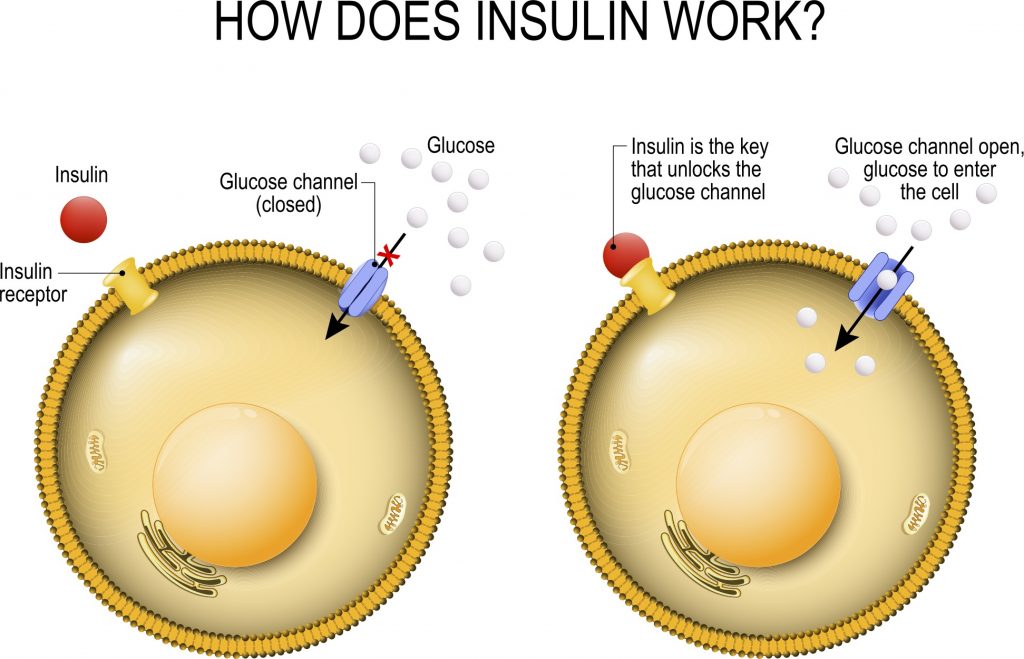
Rallentamento dello svuotamento gastrico con riduzione del senso di fame derivante dal aumento del senso di sazietà;
Gli effetti elettrofisiologici dell’attivazione del GLP-1R nelle aree cerebrali coinvolte nella modulazione del comportamento alimentare riducendo il senso di fame e il consumo di cibo;
Indirettamente, per via della stimolazione insulinica, aumento del senso di sazietà dato dal picco insulinico a livello ipotalamico;
Miglioramento del ripartizionamento Kcal attraverso il miglioramento della sensibilità all’insulina.
L'”attrattiva” che ha spinto (e che spinge) diverse persone di diverse classi sociali e di ambo i sessi a prendere in considerazione e concretizzare l’uso di questi farmaci è fondamentalmente ridotta alla riduzione della fame/appetito. Ma questo riguarda la persona “nella media”, la ragazza/donna alla ricerca di rapide soluzioni per l’imminente prova costume o per il servizio fotografico, sfilata ecc…
Nel Bodybuilding questa classe di farmaci ebbe una serie di attrattive comprendenti lo sfruttamento di tutti i punti sopra elencati. E, di conseguenza, si ipotizzarono fasi della preparazione nelle quali applicare tale categoria farmaceutica. Limitandone l’uso in “Cut” per ragioni legate al rischio (seppur limitato in monoterapia) di incorrere in eventi ipoglicemici, l'”attrattiva d’uso” riguardava il potenziale in fasi di “Recomp”; quindi non propriamente “ipocalorico” o, al massimo, leggermente ipocaloriche e non ipoglucidiche.
Ricomposizione corporea, Ripartizionamento calorico e Insulino-Sensibilità:
Ma chiariamo alcuni termini per schiarirci le idee…
Ricomposizione corporea: si intende il raggiungimento di un obiettivo o un risultato desiderato di un regime alimentare e di allenamento (e di farmaci), come il “Cut” o il “Bulk”.
“Cut”: si intende, propriamente, una diminuzione della massa grassa (FM) e il mantenimento della massa muscolare (LBM).
“Bulk”: si intende un aumento della LBM con una concomitante attenuazione dell’aumento della FM.
“Recomp” [comunemente intesa]: è definita come un aumento della LBM e la diminuzione della FM.
La suddivisione terminologica corretta è un concetto funzionale all’obiettivo di tutti gli interventi dietetici razionali.
Il rapporto p (rapporto di ripartizione) descrive le proteine depositate nei tessuti della LBM in relazione all’apporto energetico e, viceversa, le proteine perse dai tessuti della LBM in relazione al deficit energetico.
Il p-ratio comprende i fattori di:
stato ormonale (cioè i livelli assoluti di ormoni chiave noti);
sensibilità all’insulina;
sensibilità alla Leptina.
Esiste un’interazione tra il punto 1 e 3.
La prima chiave è…
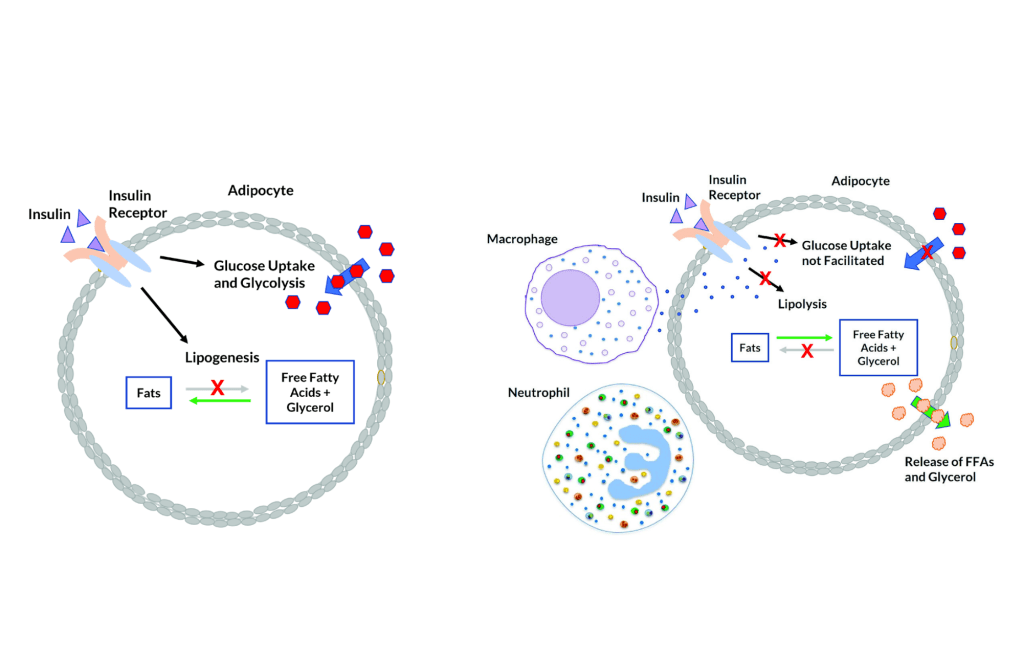
Sensibilità all’Insulina: quando si è a dieta (cioè in uno stato di deficit energetico), la resistenza all’Insulina fisiologica è una condizione favorevole all’uso del grasso di deposito limitando l’uso del glucosio da parte del muscolo come substrato energetico, risparmiando il glucosio per il cervello e l’utilizzo degli Acidi Grassi intramuscolare. In condizioni di uscita da un regime ipocalorico, in uno stato di migliorata insulino-sensibilità, l’aumento calorico di una fase di “Bulk” vede, almeno inizialmente, un ripartizionamento delle Kcal al miocita a discapito del adipocita; tale condizione di inverte col protrarsi del regime ipercalorico.
I fattori che influenzano la sensibilità all’insulina includono [1]:
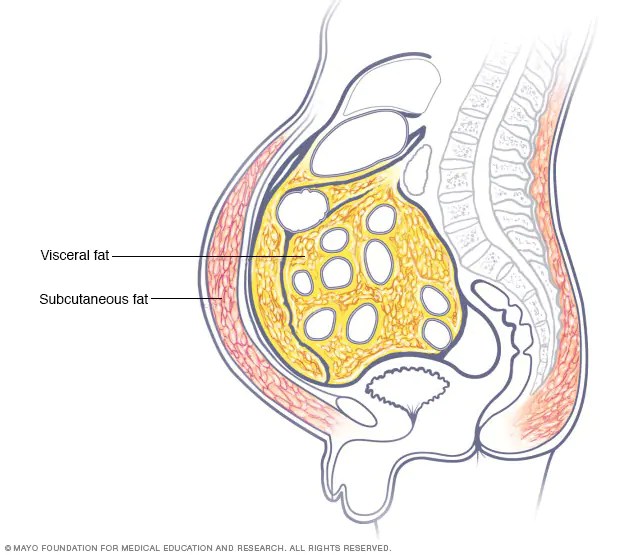
Livelli di grasso corporeo; B.F. % (predittore primario): ↑B.F. ⇒ ↑Acidi Grassi come substrato energetico (risparmiando glucosio e proteine [che possono essere utilizzate dal fegato nella gluconeogenesi]) e detta la segnalazione delle adipochine (cioè gli ormoni secernenti gli adipociti [leptina, TNF-α, IL-…, adiponectina, etc.]) ⇒ ↓ sensibilità all’insulina;
Contrazione muscolare (cioè attività, come ad esempio locomozione, allenamento contro-resistenza) ⇒ ↑ assorbimento di glucosio nella cellula muscolare; traslocazione di GLUT-4 ⇒ ↑ sensibilità all’Insulina;
Dieta: elevata quantità di carboidrati (in ipercalorica), grassi saturi e poche fibre ⇒ ↓ sensibilità all’Insulina;
Stoccaggio del Glicogeno o Supercompensazione [successivo ad una deplezione] ⇒ ↑assorbimento di glucosio e glicogenesi ⇒↑sensibilità Insulinica;
Deplezione di glicogeno (ad esempio, nel periodo successivo a un allenamento intenso, prima di un’alimentazione particolarmente ricca di carboidrati) ⇒ sottoregolazione (deplezione pressoché totale) della disponibilità di glucosio e promozione dell’ossidazione di Acidi Grassi dopo il depauperamento delle scorte di glicogeno muscolare (in media < 700 g negli adulti) ⇒ ↑ Acidi Grassi liberi nel sangue (circolanti) ⇒ ↓ sensibilità all’Insulina;
Fattori genetici in parte modificabili dai farmaci, ad esempio, nei casi di ipogonadismo, l’applicazione TRT inverte chiaramente l’insulino-resistenza nei casi in cui l’eziologia dell’insulino-resistenza è riconducibile alla carenza di Testosterone.
La seconda chiave è…
Struttura molecolare della Leptina
La Leptina: come sappiamo bene, la Leptina è un ormone, più precisamente è una adipochina, secreta principalmente dagli adipociti, che si correla con la %B.F.;↑%B.F. ⇒ ↑Leptina. (i depositi viscerali e quelli sottocutanei hanno rapporti diversi con la Leptina). A una data percentuale di B.F., le donne producono ~2 – 3 volte più Leptina rispetto agli uomini. Le concentrazioni di Leptina cambiano con la restrizione energetica e la sovralimentazione. La Leptina è un segnalatore primario di regolazione dell’accumulo di energia che riflette:
la percentuale di B.F.;
l’assunzione di energia.
Esempio 1: all’inizio di una dieta ipocalorica, la Leptina può diminuire del 50% entro 1 settimana (o meno) – anche se ovviamente il soggetto a dieta non ha perso il 50% di B.F. – quindi, in un primo momento, i cambiamenti della concentrazione di Leptina non sono correlati alla B.F. (piuttosto segnalano l’assunzione di energia).
Dopo il calo iniziale, si assiste a un declino più graduale della Leptina in relazione alla perdita di %B.F. .
Esempio 2: in caso di sovralimentazione, la Leptina subisce un incremento in modo altrettanto rapido (cioè senza relazione con la %B.F., ma in relazione all’assunzione di energia).
A breve termine, la secrezione di Leptina è determinata principalmente dalla disponibilità di glucosio, per cui la riduzione della disponibilità di glucosio nella cellula adiposa (dieta ipocalorica) ⇒ ↓Leptina e viceversa.
Gli effetti specifici dell’ormone Leptina includono effetti sul pancreas e sul fegato, nel muscolo scheletrico ↑FA e ↓AA e l’uso del glucosio come substrato energetico (aumentando la perdita di grasso, promuovendo il risparmio di proteine)… [1]
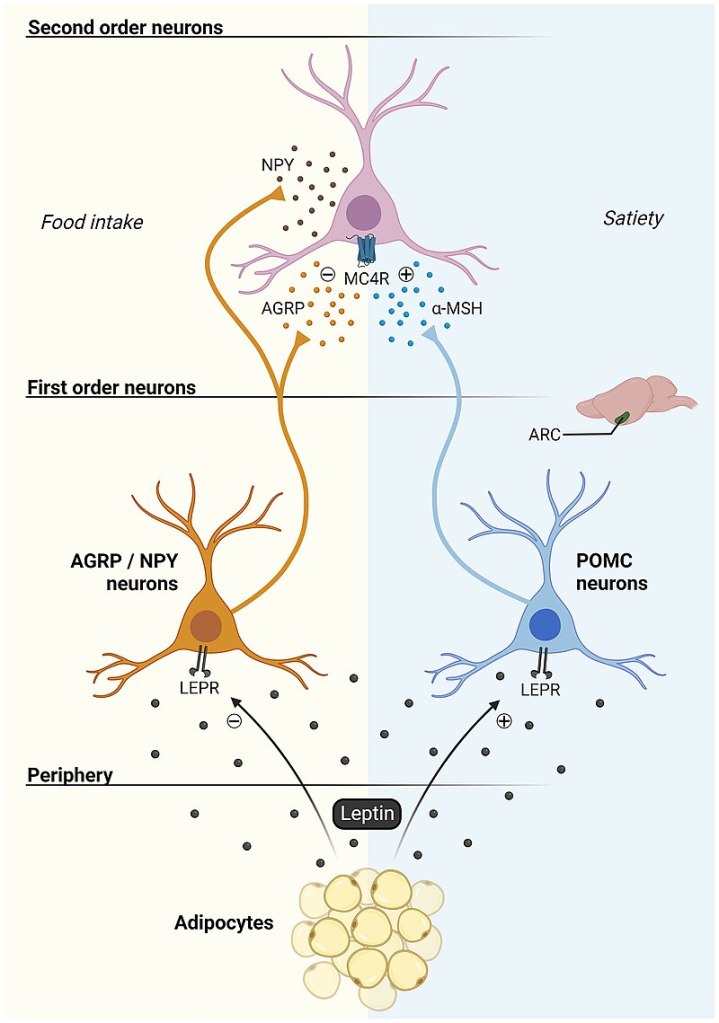
Modello classico Leptina-Melanocortina
In sostanza, il partizionamento (p-ratio) è un concetto che associa la Leptina e la sensibilità all’Insulina come fattori principali che determinano il modo in cui le variazioni dell’apporto calorico e del contenuto di macronutrienti influiscono sul metabolismo (influenzando profondamente la composizione corporea) e sullo stato ormonale. Possiamo modificarlo e migliorarlo, tenendo conto dei tessuti bersaglio e del nostro obiettivo (ad esempio, se Bulk o Cut).
Inoltre, non bisogna confondere il potenziamento dell’insulino-resistenza fisiologica sulla perdita di grasso con l’erronea valutazione che l’insulino-resistenza sia salutare. L’insulino-resistenza, soprattutto in una persona sedentaria, è associata alla sindrome metabolica, al diabete di tipo II, per non parlare del grasso viscerale, ecc.
L’insulino-resistenza è uno stato in cui i tessuti dell’organismo (ad esempio, fegato, pancreas, muscolo scheletrico) presentano una scarsa recettività con l’Insulina continuando, se si parla in particolare del fegato, a produrre glucosio in quantità inappropriate. Questo stato di iperglicemia è un effetto piuttosto che la causa dell’insulino-resistenza, anche se i livelli tossici di glucosio degradano la reattività delle isole pancreatiche all’Insulina rappresentando così una delle vie/meccanismi dell’insulino-resistenza, peggiorando la stessa condizione.
Ma tutto questo cosa centra con gli Incretino-Mimetici? Se non ci siete ancora arrivati, calma e capirete …
Agonisti del recettore GLP-1 e GIP:
La perdita di grasso si verifica con gli agonisti del GLP-1 e della GIP (Incretino-mimetici) – come la Semaglutide e la Tirzepatide – che sono veri e propri agenti sensibilizzanti dell’Insulina. Tuttavia, non è la sensibilità all’Insulina di per sé che è responsabile della perdita di grasso con questa classe di farmaci – ma piuttosto, come abbiamo già visto, la perdita di grasso avviene grazie agli altri effetti di questi farmaci, come l’alterazione potenziale delle preferenze alimentari, il ritardo dello svuotamento gastrico, il senso di sazietà, che promuovono il controllo dell’appetito e riducono l’assunzione di energia.
Sappiamo che gli agonisti del GLP-1 e del GIP migliorano direttamente la sensibilità all’insulina modulando la secrezione di Insulina – accoppiandola alla presenza di elevate concentrazioni di glucosio. Questa secrezione di Insulina si attenua quando le concentrazioni di glucosio nel sangue diminuiscono e si avvicinano all’euglicemia. Inoltre, anche se indirettamente, riducendo l’assunzione di cibo, questi farmaci determinano una riduzione della %B.F.. La riduzione della percentuale di B.F. dovuta alla riduzione dell’assunzione di cibo riduce le riserve di massa grassa (e quindi gli FFA circolanti), migliorando ulteriormente la sensibilità all’Insulina.
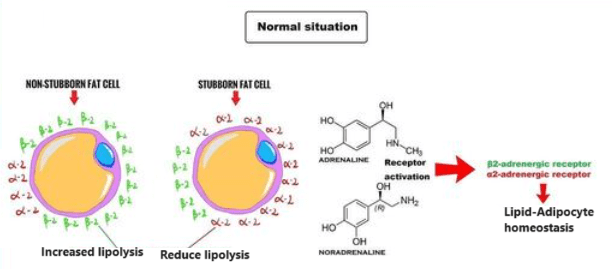
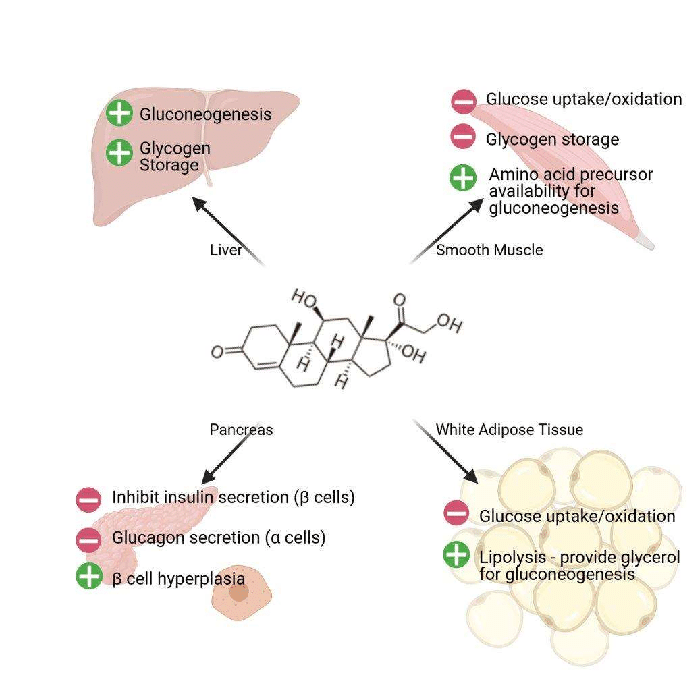
La stragrande maggioranza degli agenti per la perdita di grasso, in quanto agenti lipolitici, favoriscono l’insulino-resistenza. Ad esempio i β-agonisti, non selettivi come l’Efedrina o selettivi come il Clenbuterolo, oppure lo stimolante da banco per eccellenza la caffeina, agendo in modo analogo o aumentano l’azione delle catecolamine (epinefrina e noradrenalina, o adrenalina e noradrelanina) possono portare ad un peggioramento di questa condizione.
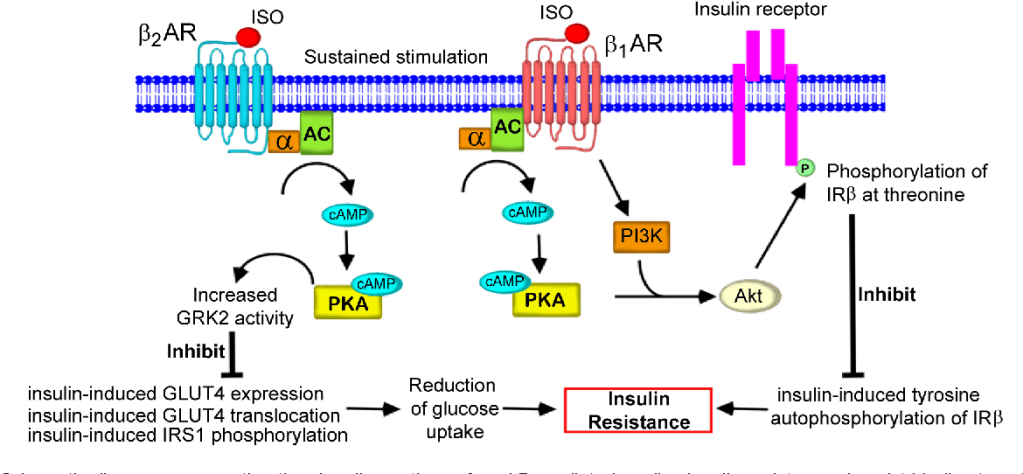
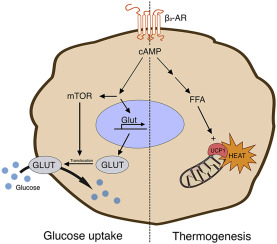
Diagramma schematico che rappresenta la via di segnalazione dell’insulino-resistenza cardiaca mediata dai β-AR. Tale meccanismo interessa (tra gli altri tessuti) anche il muscolo-scheletrico.
Quando tessuti come il fegato e le cellule adipose vedono ridotta l’interazione con l’Insulina, il glucosio non viene ottimamente assorbito dalle cellule. Con un marcato calo del glucosio, il fegato inizia a metabolizzare gli acidi grassi liberi (FFA), aumentando così i livelli di chetoni nel sangue e impedendo che vengano riesterificati nelle cellule adipose (in ipocalorica). Nel fegato e nelle cellule adipose, senza che l’Insulina interagisca ottimamente con questi tessuti, si verifica una soppressione della sintesi/lipogenesi dei grassi (negli adipociti) e della sintesi di lipoproteine a bassissima densità (VLDL) (nel fegato).
Incretino-mimetici, miglioramento della sensibilità all’Insulina e preservazione della massa muscolo-scheletrica:
La classificazione degli incretino-mimetici come agenti di ripartizione calorica:
Nel BodyBuilding, il fascino verso questa classe di farmaci si concentra anche sul funzionamento degli agonisti del GLP-1 e della GIP sull’insulino-resistenza, poiché quest’ultima durante la restrizione calorica nel muscolo scheletrico (>60% del peso corporeo, più nei bodybuilder) è un’immagine non proprio esaltante, con le riserve di glicogeno che vengono prima catabolizzate abbastanza rapidamente; poi i trigliceridi intramuscolari (che rappresentano solo l’1% del peso del muscolo idratato, fino al 2% del volume, dato che il grasso è meno denso del muscolo scheletrico, e ~1/3 dell’energia muscolare, dato che il grasso è energeticamente denso) e infine, se necessario, l’organismo utilizzerà gli AA (catabolizzando le proteine muscolari; proteolisi) per ottenere l’energia necessaria a svolgere le attività giornaliere. Questi agenti, quindi, nella misura in cui sono sensibilizzanti per l’Insulina, dovrebbero servire a promuovere il mantenimento della LBM durante il “Cut”.
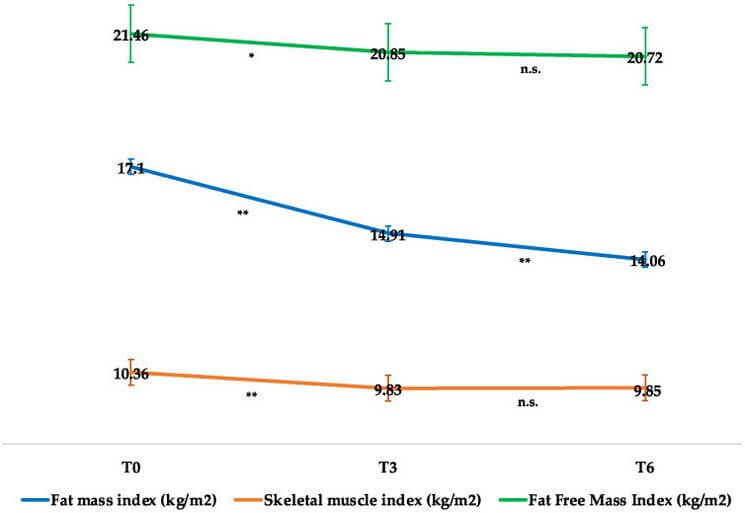
In effetti, come si evince dalla seguente immagine tratta da un articolo di ricerca di Volpe et. al del 2022 [2], la Semaglutide preserva in modo abbastanza efficace la LBM e riduce in modo preferenziale la FM, con riduzioni solo clinicamente insignificanti dell’indice di massa magra (FFMI, kg/m²) e dell’indice della muscolatura scheletrica durante il periodo iniziale di adattamento, che poi si attenua:
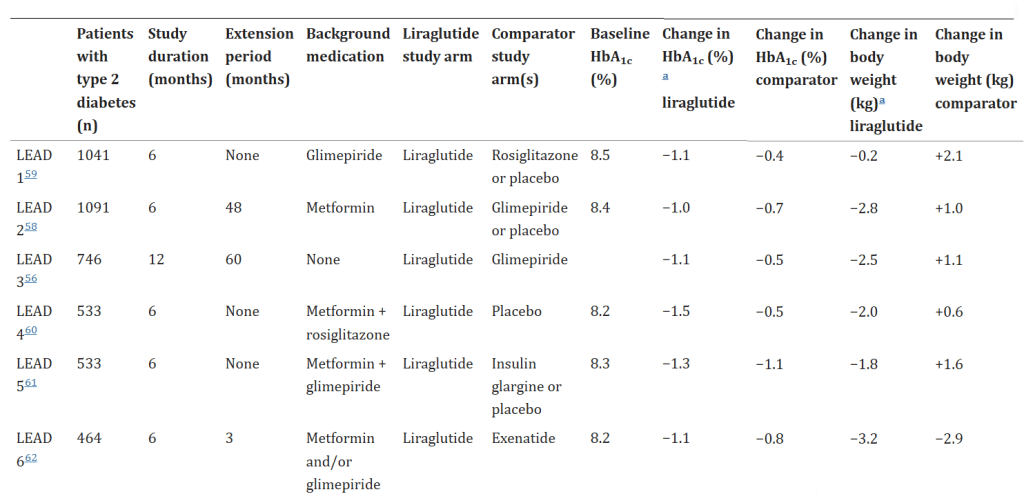
In un certo senso, quindi, migliorando il grado di sensibilità all’Insulina dell’equazione della p-ratio, gli incretino-mimetici possono essere classificati come agenti di ripartizione calorica, a grandi linee come il Clenbuterolo, ma invece di promuovere l’insulino-resistenza come i β-agonisti, la migliorano.
* Ovviamente, creare un ambiente significativamente insulino-sensibile in un contesto ipocalorico (soprattutto se ipoglucidico) può mettere l’utilizzatore a maggior rischio (sebbene limitato) di ipoglicemia o calo glicemico borderline con effetti simili allo stato di ipoglicemia (tremore, sudorazione copiosa ecc…).
Per coloro che hanno familiarità con questi concetti, derivanti dalle discussioni sul bodybuilding, può risultare molto confuso il fatto che l’iperglicemia (elevata quantità di glucosio nel sangue) è solo uno dei fattori associati all’insulino-resistenza, ma in realtà non è sinonimo di insulino-resistenza (iperglicemia ≠ insulino-resistenza). Sì, ridurre la glicemia a livelli normali è molto importante per migliorare la sensibilità all’insulina durante l’uso dell’ormone della crescita esogeno (rhGH), perché il glucosio è tossico per le cellule β pancreatiche. Questa glucotossicità a livello delle cellule pancreatiche si traduce in una diminuzione della risposta secretoria dell’insulina all’iperglicemia, alimentando così il fuoco dell’iperglicemia e della glucotossicità, contribuendo all’insulino-resistenza – ma non costituendone l’unica eziologia.
La sensibilità all’Insulina è multifattoriale e comprende componenti sistemiche (ad esempio, QUICKI) e periferiche (ad esempio, GLUT-4) ed è regolata a livello centrale da GLP-1 e GIP. L’iperglicemia è solo uno dei fattori (l’altro è l’Insulina) che funge da proxy dell’insulino-resistenza sistemica. Vi sono altri aspetti, tra cui la tolleranza ai carboidrati [vedi anche capacità di tolleranza del metabolismo glucidico], ecc.
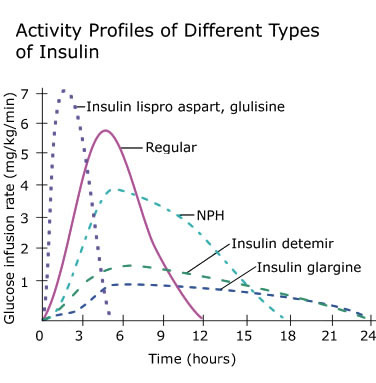
In tema di insulino-resistenza, è utile ricordare che l’Insulina endogena viene secreta in modo pulsatile per regolare il metabolismo glucidico e lipidico, la crescita cellulare ecc…, a differenza del Testosterone che viene secreto in modo più stabile (rilascio graduale nel sangue, ma soggetto a variazioni diurne, ad esempio una maggiore secrezione al mattino rispetto a mezzogiorno/sera). Gli aumenti cronici di Insulina, ad esempio quelli relativi al profilo di rilascio di una bassa dose giornaliera di Insulina Glargine (Lantus), presentano un’area sotto la curva (AUC) relativamente ampia a causa del profilo di rilascio (concentrazioni elevate per lunghi periodi di tempo) rispetto ai normali profili di rilascio dell’Insulina endogena (paragonabili alla farmacocinetica dell’Insulina regolare, ad esempio Actrapid, Novolin o HumuLin -R). L’elevata AUC di Lantus e/o le dosi di Insulina regolare esogena moderatamente elevate e frequenti sono descritte come iperinsulinemia cronica.
Questa resistenza non avviene per feedback negativo a livello delle cellule β.
Al contrario, l’iperinsulinemia cronica che causa l’insulino-resistenza è multifattoriale e comprende:
L’aumento dell’HOMA-IR e la diminuzione del QUICKI (misure biochimiche dell’insulino-resistenza e della sensibilità all’Insulina, rispettivamente).
L’alterata trasduzione del segnale insulinico dovuta alla disfunzione del recettore dell’Insulina (IR) e alla diminuzione dell’autofosforilazione dell’IR, che blocca la traslocazione del GLUT-4 sulla superficie cellulare nelle cellule muscolari e adipose, con conseguente aumento del glucosio nel sangue [3]:
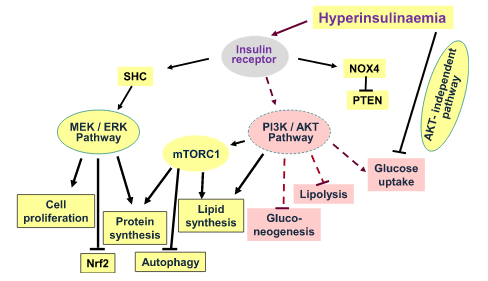
Segnalazione dell’Insulina durante l’insulino-resistenza. Durante l’insulino-resistenza, la segnalazione attraverso le chinasi AKT è parzialmente compromessa. Non tutte le vie AKT-dipendenti sono interessate, così come altre vie di segnalazione, indicando che l’insulino-resistenza è selettiva. Pertanto, l’iperinsulinemia, in presenza di insulino-resistenza, promuove le attività anaboliche delle cellule attraverso la via MEK-ERK e attraverso mTORC1. Sebbene la via PIK/AKT sia compromessa durante l’insulino-resistenza e fornisca solo una traslocazione insufficiente di GLUT4 per l’assorbimento del glucosio e un’attivazione carente di eNOS, sembra esserci un’attivazione normale di mTORC1. Oltre alle conseguenze anaboliche della segnalazione attraverso la via MEK/ERK descritte nella figura, si osserva un’aumentata espressione di ET-1 e PAt-1 (non mostrato), nonché l’inibizione dell’autofagia e del fattore nucleare Nrf2, che compromettono rispettivamente il turnover dei costituenti cellulari e i meccanismi di difesa delle cellule dallo stress radicale. L’iperinsulinemia riduce l’assorbimento del glucosio non solo attraverso lo smorzamento della via PIK/AKT (resistenza all’Insulina), ma anche attraverso altre vie ancora sconosciute.
3. Aumento dei livelli e dell’attività di sn-1,2-diacilglicerolo (DAG) grazie alla sintesi de novo.
Le limitazioni degli Incretino-Mimetici dietro all’iniziale entusiasmo:
Se ci dovessimo basare su quanto esposto fino a questo punto, saremo tutti d’accordo nell’ammantare della nomea di “farmaci prodigiosi per la ricomposizione corporea” tanto la Semaglutide quanto il Tirzepatide e tutti gli altri membri di questa classe. Ma, dal momento che, la conoscenza per essere utile deve essere sufficientemente approfondita, occorre indagare meglio sulle caratteristiche di questi farmaci.
Sappiamo che la Semaglutide è effettivamente associata alla perdita di peso con una differenza media dell’11,85% rispetto al placebo emersa dalle ultime review. Il consolidamento degli studi ha mostrato che nausea, vomito, costipazione e diarrea sono gli eventi avversi più comuni. Nonostante questi effetti sembrano essere per lo più di gravità da lieve a moderata, la loro risoluzione totale era spesso connessa al termine del trattamento.
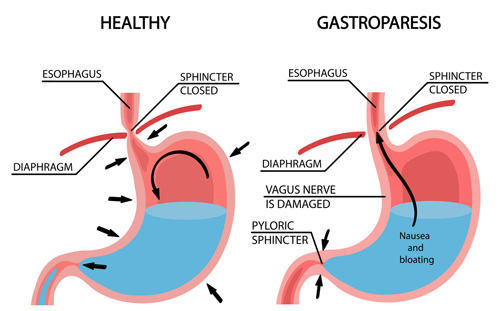
Dal momento che gli Incretino-mimetici causano una riduzione del senso di fame, in modo anche significativo (dose dipendente), rallentano lo svuotamento gastrico portando anche ad eventi diarroici preceduti da mal assorbimento dei nutrienti, senza sottovalutare il rischio di paresi gastrica, la facilità di perdere massa muscolare in un contesto ipocalorico è molto alta, specie se non supportata da agenti anabolizzanti; l’inserimento di questi ultimi, però, non “tampona” la possibile condizione di mal assorbimento e/o scarsa assunzione proteica/alimentare.
Inoltre, la paresi gastrica è una condizione che un bodybuilder sano di mente cerca di evitare a tutti i costi combattendoci già se sussiste abuso di Insulina e/o hGH.
Trattando la limitazione data da una potenziale eccessiva inappetenza, è giusto specificare un impatto singolare che le Incretine (e gli Incretino-Mimetici) hanno sulle preferenze alimentari. Sappiamo, infatti, che gli alimenti maggiormente palatabili sono tipicamente ricchi di grassi e/o zuccheri e tendono a essere preferiti a quelli a basso contenuto di grassi/zuccheri. L’entità di questa preferenza, tuttavia, può essere influenzata da peptidi intestinali quali la Grelina e il GLP-1. La Grelina e il GLP-1 sono influenzati in modo differenziato dal consumo di alimenti palatabili. Tralasciando la Grelina, le concentrazioni di GLP-1 a digiuno predicono negativamente l’assunzione di alimenti ricchi di zuccheri semplici in un paradigma di distributori automatici, che gli autori dello studio hanno interpretato come prova del fatto che il GLP-1 svolge un ruolo nelle vie di ricompensa che regolano l’assunzione di zuccheri semplici. Diversi studi riportano anche un’alterazione delle preferenze alimentari dopo l’intervento di bypass gastrico, con un allontanamento dalla preferenza per gli zuccheri/grassi elevati. L’assunzione di alimenti appetibili, in particolare di soluzioni zuccherate, è aumentata dalla Grelina, mentre il GLP-1 riduce preferenzialmente l’assunzione di alimenti ad alto contenuto di grassi e zuccheri, almeno dopo una somministrazione acuta. Inoltre, i lavori condotti sull’uomo rivelano che la preferenza per i grassi e gli zuccheri può essere alterata dalla chirurgia bariatrica e contribuire alla perdita di peso, ma non è ancora stato stabilito se questi effetti siano legati a un’alterazione del segnale della Grelina o del GLP-1. Infine, i livelli circolanti di Grelina e GLP-1 possono essere indicativi del consumo di cibo appetibile nell’uomo.[https://www.frontiersin.org/]
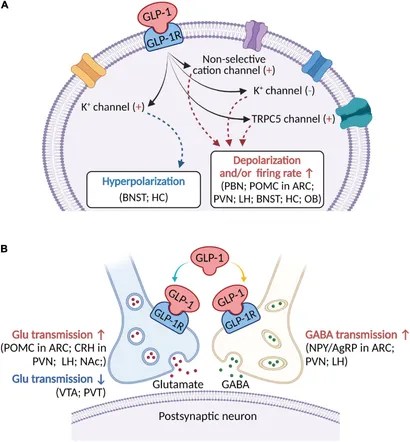
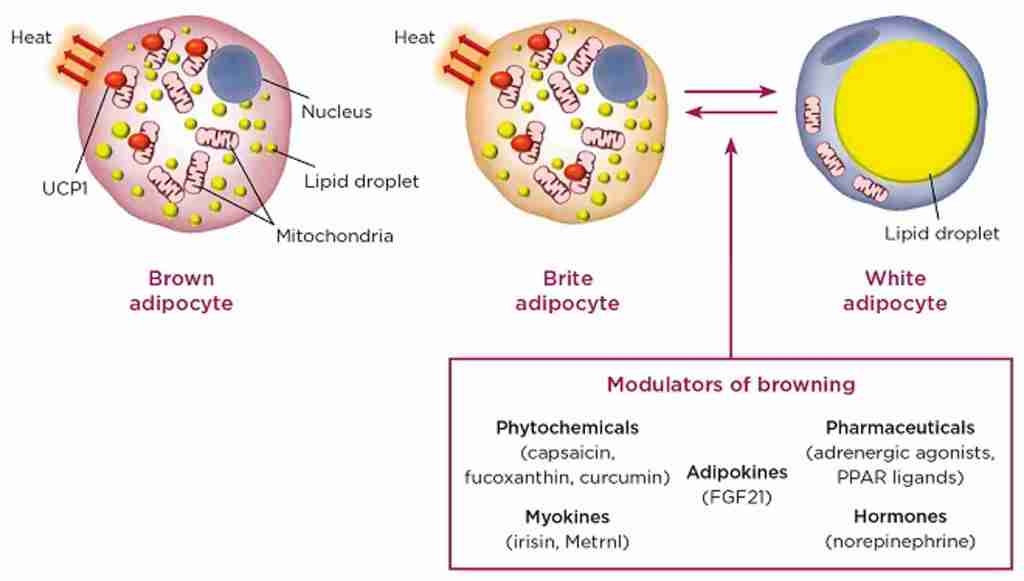
Schema che descrive i principali effetti elettrofisiologici dell’attivazione del GLP-1R nelle aree cerebrali coinvolte nella modulazione del comportamento alimentare. (A) Il GLP-1 (compresi i suoi agonisti) si lega al GLP-1R postsinaptico per depolarizzare il potenziale di membrana e/o aumentare la frequenza di sparo nella maggior parte delle regioni cerebrali, ma iperpolarizza il potenziale di membrana in alcune aree cerebrali. Diversi meccanismi ionici, tra cui il canale cationico non selettivo, il canale K+ e il canale TRPC5, possono essere coinvolti nell’attivazione della depolarizzazione o iperpolarizzazione indotta dal GLP-1R. (B) Oltre ai recettori postsinaptici, il GLP-1 agisce sui GLP-1R presinaptici per modulare la neurotrasmissione glutammatergica e GABAergica. ARC, nucleo arcuato; BNST, nucleo del letto della stria terminale; Glu, glutammato; CRH, ormone di rilascio della corticotropina; HC, ippocampo; LH, ipotalamo laterale; NAc, nucleo accumbens; NPY/AgRP, Neuropeptide Y/Peptide legato al gene Agouti; OB, bulbo olfattivo; PBN, nucleo parabrachiale; POMC, proopiomelanocortina; PVN, nucleo paraventricolare; PVT, nucleo talamico paraventricolare; VTA, area tegmentale ventrale.
Di conseguenza, nonostante gli indizi di cui sopra, l’uso di Incretino-Mimetici potrebbe ridurre marcatamente il consumo di cibo indipendentemente dalla fonte anche se, come abbiamo visto, l’effetto anoressizzante del GLP-1 sembra essere a maggior carico della componente alimentare glucidico-lipidica.
Tornando invece sulla questione legata al catabolismo muscolo-scheletrico e l’uso concomitante di agenti anabolizzanti, per ovviare a questo problema nei pazienti trattati con Semaglutide, è stato avviato uno studio clinico di fase 2b, multicentrico, in doppio cieco, controllato con placebo, randomizzato, per la determinazione della dose e per valutare la sicurezza e l’efficacia di Ostarina 3mg, Ostarina 6mg o placebo come trattamento per preservare la massa muscolare e aumentare la perdita di grasso in circa 150 pazienti con obesità sarcopenica o pazienti anziani in sovrappeso (>60 anni di età) trattati con Semaglutide (Wegovy®). L’endpoint primario è la massa corporea magra totale e gli endpoint secondari chiave sono la massa grassa corporea totale e la funzione fisica misurata dal test di salita delle scale a 16 settimane.
Dopo aver completato la parte di determinazione della dose di efficacia dello studio clinico di Fase 2b, si prevede che i partecipanti continueranno in cieco in uno studio clinico di estensione di Fase 2b in cui tutti i pazienti smetteranno di ricevere un GLP-1 RA, ma continueranno ad assumere un placebo, Ostarina 3mg o Ostarina 6mg per ulteriori 12 settimane. Lo studio clinico di estensione di fase 2b valuterà se l’Ostarina è in grado di mantenere la massa muscolare e prevenire l’aumento di grasso e peso che si verifica dopo l’interruzione di un GLP-1 RA.
Lo studio clinico è condotto in 14 centri clinici negli Stati Uniti. È stato raggiunto l’arruolamento completo dei circa 150 pazienti nello studio di fase 2b QUALITY. L’azienda prevede ora che l’ultimo paziente a completare lo studio di fase 2b QUALITY sarà nel dicembre 2024, con i risultati clinici di prima linea per lo studio clinico di fase 2b QUALITY attesi nel gennaio 2025. Inoltre, i risultati principali per lo studio clinico di estensione di Fase 2b in cieco separato possono ora essere attesi nel secondo trimestre solare del 2025.
Ovviamente, queste limitazioni, incisive nel Culturismo, interessano tutti gli Incretino-Mimetici, compresa la Tirzepatide la quale sembrerebbe avere un maggior margine di efficacia e “sicurezza” per quanto riguarda il rischio (seppur limitato in monoterapia) di eventi ipoglicemici.
Il motivo di questa riduzione di rischio ipoglicemico è dovuta alla doppia affinità recettoriale della Tirzepatide la quale, come abbiamo visto, possiede una attività agonista per i recettori del GLP-1 e del GIP. Ed è proprio il legame e l’attivazione di quest’ultimo recettore (GIP) da parte della Tirzepatide a permettere ciò. Infatti, l’attivazione del recettore GIP stimola la secrezione di Glucagone in modo glucosio-dipendente nelle persone sane, con un’attività maggiore in presenza di glicemie più basse. Ciò significa che, raggiunta la soglia euglicemica i livelli di glucosio nel sangue verranno mantenuti più facilmente all’interno di questa soglia per via dell’attività del Glucagone secreto dalle cellule α delle isole di Langerhans.[https://pubmed.ncbi.nlm.nih.gov/]
Sebbene i trials clinici suggeriscano che la Tirzepatide riduca la glucagonemia, un recente studio dimostra che la Tirzepatide è un potente stimolatore della secrezione di Glucagone nelle condizioni sopradette.
Quindi la Tirzepatide è superiore alla Semaglutide?
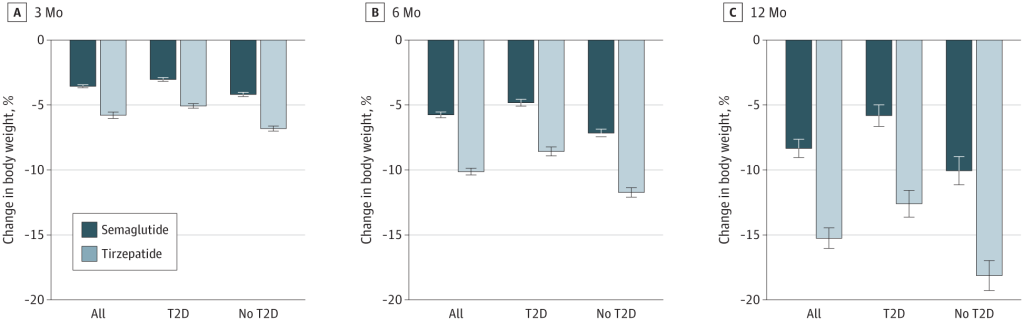
Alcuni studi recenti hanno messo a confronto la Semaglutide e la Tirzepatide per la perdita di peso. Studi di ricerca del 2021, del 2023 e del 2024 suggeriscono che la Tirzepatide può determinare una maggiore perdita di peso rispetto alla Semaglutide.
Variazione percentuale media del peso corporeo a 3, 6 e 12 mesi di trattamento per la popolazione complessiva, i soggetti con diabete di tipo II (TD2) e quelli senza TD2. Le barre rappresentano le variazioni medie del peso corporeo dal basale al punto di riferimento tra la popolazione di pazienti ancora in trattamento, abbinata in base al punteggio di propensione. Barre scure (Semaglutide); Barre chiare (Tirzepatide).
Ma questi studi presentano alcuni importanti limiti.
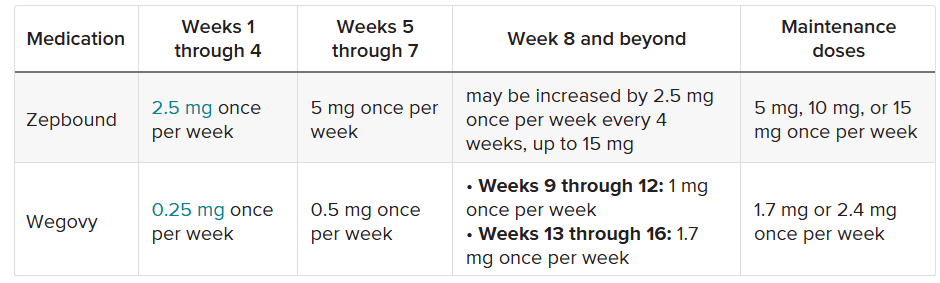
Le dosi di Semaglutide e Tirzepatide somministrate ai partecipanti non erano, per ovvie ragioni, uguali. La Semaglutide e Tirzepatide funzionano in modo leggermente diverso, come ormai sappiamo, e quindi i ricercatori hanno scelto livelli di dose comparabili. Tuttavia, la dose di Tirzepatide era più alta, il che potrebbe aver, anche di poco, influenzato i risultati.
Tabella comparativa tra il dosaggio della Semaglutide [Wegovy] e quella della Tirzepatide [Zepbound] usato negli studi.
Sappiamo altresì che la Tirzepatide ha una maggiore affinità con i recettori GIP rispetto ai recettori GLP-1. Di conseguenza, la ratio della dose di Tirzepatide con quella di Semaglutide risulta maggiormente a carico della prima.
La Tirzepatide è attualmente approvata dalla FDA per l’uso in persone in sovrappeso o con obesità, indipendentemente dal fatto che soffrano o meno di diabete di tipo II. Tuttavia, alcuni studi suggeriscono che la Tirzepatide sia un farmaco che non ha bisogno di essere somministrato in caso di mancanza della condizione diabetica.
Negli studi dove la Tirzepatide è stata somministrata a soggetti obesi, sono comunque stati osservati miglioramenti in tutte le misure cardiometaboliche. Gli eventi avversi più comuni con la Tirzepatide sono i medesimi riscontrati con la Semaglutide o altri membri della stessa famiglia. Essi sono stati di tipo gastrointestinale e la maggior parte di questi sono stati di gravità lieve o moderata e si sono verificati principalmente durante l’aumento della dose. Gli eventi avversi hanno causato l’interruzione del trattamento nel 4,3%, 7,1%, 6,2% e 2,6% dei partecipanti che hanno ricevuto dosi di Tirzepatide da 5, 10 e 15mg e placebo, rispettivamente.
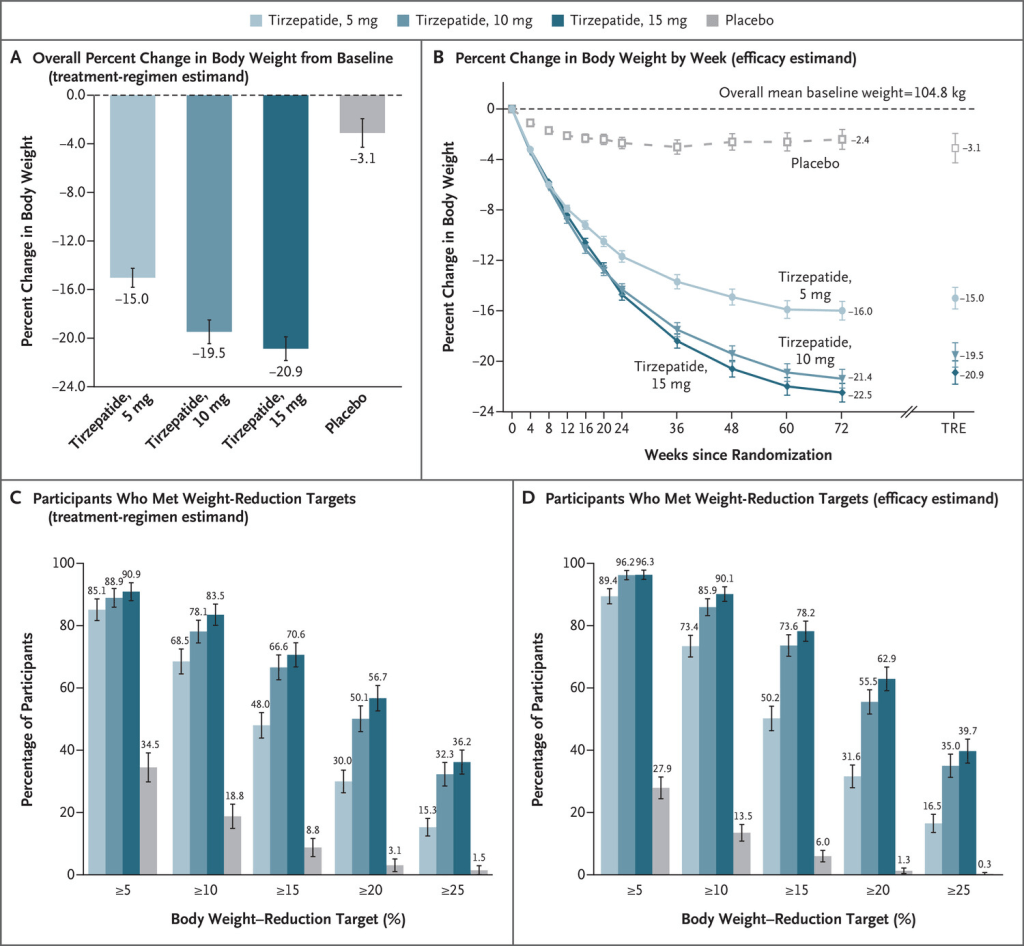
In uno studio di 72 settimane su partecipanti con obesità, 5mg, 10mg o 15mg di Tirzepatide una volta alla settimana hanno fornito riduzioni sostanziali e durature del peso corporeo.
Effetto della Tirzepatide somministrata una volta alla settimana, rispetto al placebo, sul peso corporeo. Le medie dei minimi quadrati sono presentate, se non diversamente specificato. Il pannello A mostra la variazione percentuale del peso corporeo dal basale alla settimana 72, derivata da un modello di analisi della covarianza per la stima del regime di trattamento (TRE). Il pannello B mostra la variazione percentuale del peso corporeo in base alle settimane dalla randomizzazione, derivata da un modello misto per misure ripetute (MMRM) per la stima dell’efficacia; sono riportate anche le stime alla settimana 72 per la stima del regime di trattamento. I riquadri C e D mostrano le percentuali di partecipanti che hanno avuto riduzioni di peso di almeno il 5%, 10%, 15%, 20% e 25% dal basale alla settimana 72. Per il riquadro C, la percentuale è stata calcolata in base al numero di settimane di randomizzazione. Per il pannello C, la percentuale è stata calcolata con l’uso delle regole di Rubin, combinando le percentuali dei partecipanti che hanno raggiunto l’obiettivo nei set di dati imputati. I valori mancanti alla settimana 72 sono stati imputati con MMRM se la mancanza era dovuta esclusivamente a Covid-19 e con imputazione multipla se la mancanza non era dovuta a Covid-19. Per il pannello D, la percentuale di partecipanti che hanno raggiunto gli obiettivi di riduzione del peso è stata ottenuta dividendo il numero di partecipanti che hanno raggiunto i rispettivi obiettivi alla settimana 72 per il numero di partecipanti con un valore al basale e almeno un valore post-base non mancante. I valori mancanti alla settimana 72 sono stati imputati dall’analisi MMRM. Le barre 𝙸 indicano gli intervalli di confidenza al 95%.
Nella pratica su bodybuilder, si sono osservati i minori sides comparati a ottimi risultati su insulino-resistenza e riduzione contenuta della fame con dosaggi settimanali di 2.5mg.
Alla luce di ciò, e in contesto aspecifico, la Tirzepatide mostra un moderato vantaggio gestionale rispetto alla Semaglutide.
Si è parlato di un ipotetico “rebound” di Grelina con incremento della fame e del consumo calorico (con aumento di peso) dopo la cessazione d’uso di Semaglutide o Tirzepatide. Al momento non esistono dati certi che ci indichino un reale collegamento equazionale tra cessazione d’uso di GLP-1 agonisti > picco in cronico della Grelina > iperfagia > aumento ponderale di peso. Sappiamo, però, che le variazioni di Grelina e GLP-1 a 6 mesi dalla cessazione di una dieta ipocalorica non hanno predetto il recupero del peso da 6 a 18 mesi. Ciò significa che, in un soggetto sano, potrebbe si esserci una maggiore attività della Grelina nelle prime settimane dopo cessazione d’uso di un incretino-mimetico (calo soglia ematica del farmaco e stabilizzazione dei livelli endogeni di GLP-1), ma l’aumento del peso successivo potrebbe risultare con maggiore probabilità dalla modifica omeostatica multi-fattoriale la quale, per trovare un nuovo equilibrio, richiede per lo meno un arco di tempo direttamente proporzionale al tempo di trattamento. Si consideri, inoltre, che un anno dopo la sospensione della Semaglutide sottocutanea a 2,4mg una volta alla settimana e dell’intervento sullo stile di vita, i soggetti possono mostrare una riacquisizione di due terzi della perdita di peso precedente, con cambiamenti simili nelle variabili cardiometaboliche. Qualcosa di un possibile rebound grelinico…
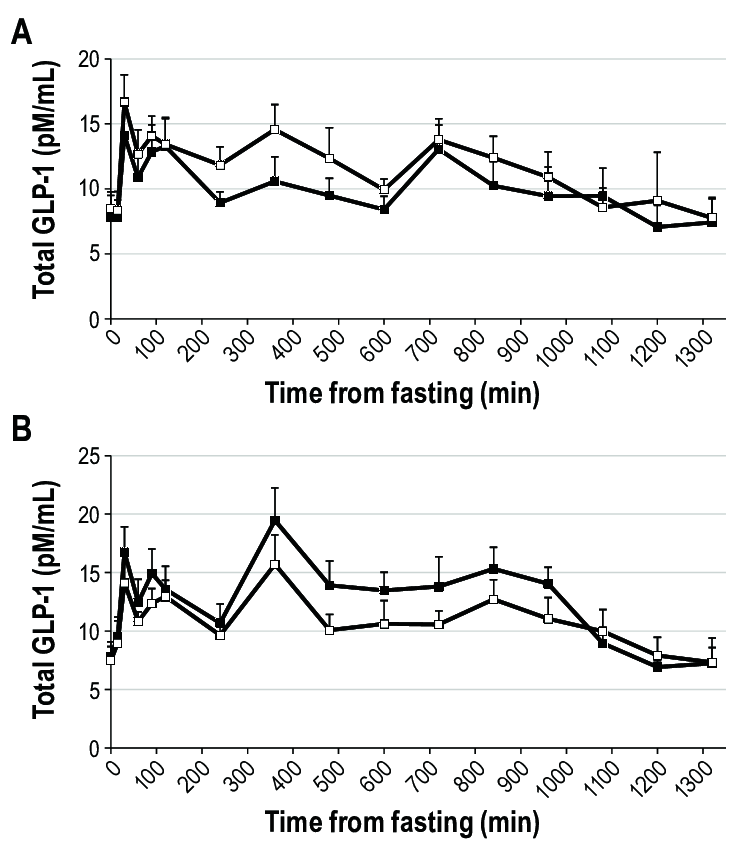
Concentrazione totale di GLP-1 durante un giorno di frequenti prelievi di sangue dopo 3 notti di sonno abituale (9 ore a letto, riquadri neri) o breve (4 ore a letto, riquadri bianchi) in uomini (pannello A) e donne (pannello B). Il tempo è presentato come minuti dal campione a digiuno. Il campione a digiuno è stato prelevato alle 08:00. I pasti e gli spuntini sono stati serviti dopo i prelievi di 0, 240 e 480 minuti e a 660 minuti. L’ora di andare a letto era a 840 minuti (sonno abituale) e a 1.020 minuti (sonno breve) rispetto al prelievo a digiuno (equivalente alle 22:00 e alle 01:00 per il sonno abituale e breve, rispettivamente). I livelli mattutini (P = 0,10) e notturni (P = 0,12) tendevano a essere più bassi e i livelli pomeridiani erano significativamente più bassi (P = 0,016) durante il sonno breve rispetto al sonno abituale nelle donne, mentre negli uomini le concentrazioni pomeridiane di GLP-1 tendevano a essere più alte dopo il sonno breve rispetto al sonno abituale (P = 0,10). I dati sono medie non aggiustate e SEM, n = 14 uomini o 13 donne.Concentrazione di Grelina totale durante un giorno di frequenti prelievi di sangue dopo 3 notti di sonno abituale (9 ore a letto, riquadri neri) o breve (4 ore a letto, riquadri bianchi) in uomini (pannello A) e donne (pannello B). Il tempo è presentato come minuti dal campione a digiuno. Il campione a digiuno è stato prelevato alle 08:00. I pasti e gli spuntini sono stati serviti dopo i prelievi di 0, 240 e 480 minuti e a 660 minuti. L’ora di andare a letto era a 840 minuti (sonno abituale) e a 1.020 minuti (sonno breve) rispetto al prelievo a digiuno (equivalente alle 22:00 e alle 01:00 per il sonno abituale e breve, rispettivamente). I livelli di Grelina mattutina più elevati sono stati osservati dopo il sonno breve rispetto al sonno abituale negli uomini; nelle donne non sono state osservate differenze tra i periodi di sonno. I dati sono medie non aggiustate e SEM, n = 14 uomini o 13 donne.
Conclusioni:
Arrivati alla conclusione di questa disamina abbiamo tutti gli elementi per valutare l’eventuale senso di utilizzo degli Incretino-Mimetici in contesti al di fuori del trattamento del diabete di tipo II o di soggetti obesi.
Le limitazioni date dagli effetti collaterali più comuni, se contestualizzate in una preparazione di un bodybuilder, possono causare non poche problematiche specie nelle vicinanze di una gara; vedi, ad esempio, estrema riduzione dell’assunzione calorica e proteica, mal assorbimento e gonfiore addominale o paresi gastrica.
Sebbene la possibilità di eventi ipoglicemici sia contenuta, e ancor più rara con la Tirzepatide in monoterapia, il rischio, in concomitanza dell’effetto sulla insulino-sensibilità degli AAS o di altre molecole co-somministrate, di questo sides può aumentare in modo sensibile durante una dieta ipocalorica.
In tal sede non ho preso in considerazione i due più preoccupanti, e più rari, effetti collaterali legati all’uso di incretino-mimetici: Tumore Midollare della Tiroide [MTC] e Pancreatite. Quest’ultima può manifestarsi anche con l’uso di AAS, seppur raramente, specie in caso d’uso di molecole aromatizzabili; la presenza di un incretino-mimetico in tali circostanze potrebbe avere un incidenza maggiore nello sviluppo e manifestazione della Pancreatite.[https://jmedicalcasereports.biomedcentral.com/]
In definitiva, i vantaggi potenziali di una una insulino-sensibiltà maggiore iatrogeno-dipendente (visti in precedenza) con l’uso di Incretino-Mimetici è, con i dovuti distinguo complessivi, ottenibile con l’uso di Metformina la quale presenta un margine di sicurezza decisamente più ampio.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Semaglutide and Tirzepatide are More Than Just Weight Loss Drugs [di Type-IIx]
[1] McDonald, L. The Ultimate Diet 2.0: Advanced Cyclical Dieting for Achieving Super Leanness. (2003). Lyle McDonald Publishing.
[2] Volpe S, Lisco G, Racaniello D, Fanelli M, Colaianni V, Vozza A, Triggiani V, Sabbà C, Tortorella C, De Pergola G, Piazzolla G. Once-Weekly Semaglutide Induces an Early Improvement in Body Composition in Patients with Type 2 Diabetes: A 26-Week Prospective Real-Life Study. Nutrients. 2022 Jun 10;14(12):2414. doi: 10.3390/nu14122414.
[3] Kolb H, Kempf K, Röhling M, Martin S. Insulin: too much of a good thing is bad. BMC Med. 2020;18(1):224. Published 2020 Aug 21. doi:10.1186/s12916-020-01688-6
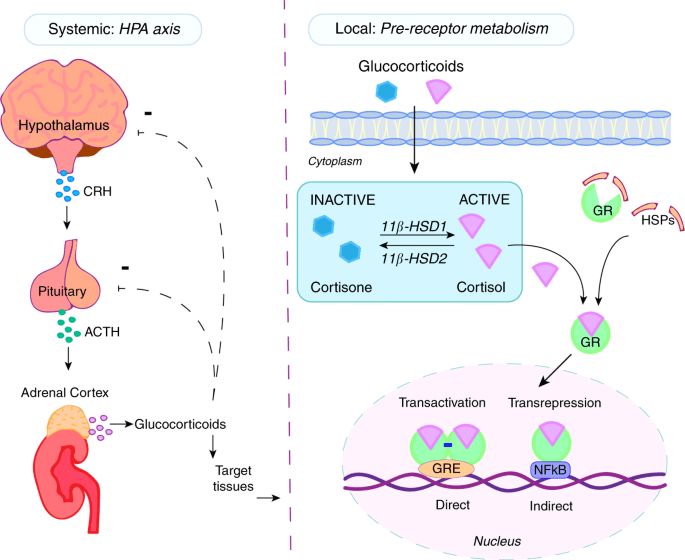
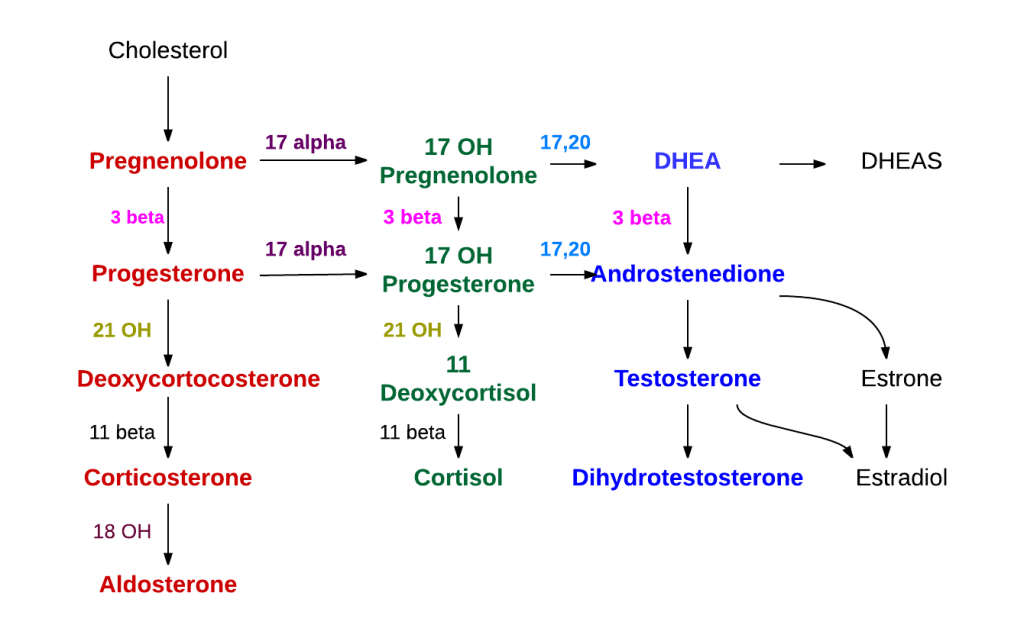
Gli incretino-mimetici sono una recente classe di farmaci antidiabete che prevede la modulazione del sistema delle Incretine. Si legano e attivano i recettori del peptide glucagone-simile-1 (GLP-1) sulle beta-cellule pancreatiche, dando inizio alla secrezione e alla sintesi di Insulina. Gli incretino-mimetici di ultima generazione hanno attività sia sul recettore del GLP-1 sia su quello del Polipeptide Inibitore Gastrico (GIP), un ormone inibitore della famiglia delle secretine (Incretine) con ruolo principale, essendo un’Incretina, di stimolare la secrezione di Insulina.
Poiché questi composti non hanno attività insulinotropica a concentrazioni di glucosio inferiori, il rischio di ipoglicemia – una nota carenza degli attuali trattamenti antidiabete – è basso. Inoltre, è stato dimostrato che gli incretino-mimetici sono associati a effetti benefici sui fattori di rischio cardiovascolare, come la perdita di peso, la riduzione della pressione sanguigna e le modifiche del profilo lipidico. Ciò nonostante, gli effetti avversi che possono causare non sono trascurabili e la loro diffusione per uso “Off-Label” ne ha mostrato le limitazioni sia in soggetti obesi che in utilizzatori per finalità estetiche.
La disamina che seguirà sarà divisa in due parti preposte a spiegare, a partire dalla loro genesi, il funzionamento delle incretine per poi passare in rassegna le caratteristiche di quelle che sono le forme farmaceutiche di incretino-mimetici utilizzate in medicina e in ambito “Off-Label/ricreativo”.
In questa prima parte vedremo nel dettaglio le incretine endogene e le molecole incretino-mimentiche presenti nel mercato…
Ormoni Incretinici [Incretine]:
Joel Habener
Negli anni ’70, Jens Juul Holst e Joel Habener iniziarono la ricerca sull’ormone GLP-1, inizialmente in relazione alla malattia dell’ulcera duodenale.[1] Esaminarono gli ormoni secreti durante l’alimentazione e li testarono su pancreas di maiale, portando alla scoperta della notevole potenza del GLP-1 nel 1988. Il loro lavoro, che in seguito ha contribuito in modo significativo al trattamento del diabete e dell’obesità, è valso a loro e a Daniel Drucker il premio della Fondazione Warren Alpert del 2021.[1] La ricerca è proseguita e nel 1993 Michael Nauck è riuscito a infondere il GLP-1 in persone affette da diabete di tipo II, stimolando l’Insulina e inibendo il Glucagone e portando la glicemia a livelli normali. Tuttavia, il trattamento dei pazienti diabetici con gli ormoni GLP-1 ha provocato notevoli effetti collaterali, inducendo i ricercatori finanziati da Novo Nordisk a cercare di sviluppare un composto adatto all’uso terapeutico.[1]
Nel 1998 un team di ricercatori di Novo Nordisk, guidato dalla scienziata Lotte Bjerre Knudsen, ha sviluppato la Liraglutide, un agonista del recettore del peptide glucagone-simile-1 con potenziale di utilizzo per il trattamento del diabete di tipo II.[2]
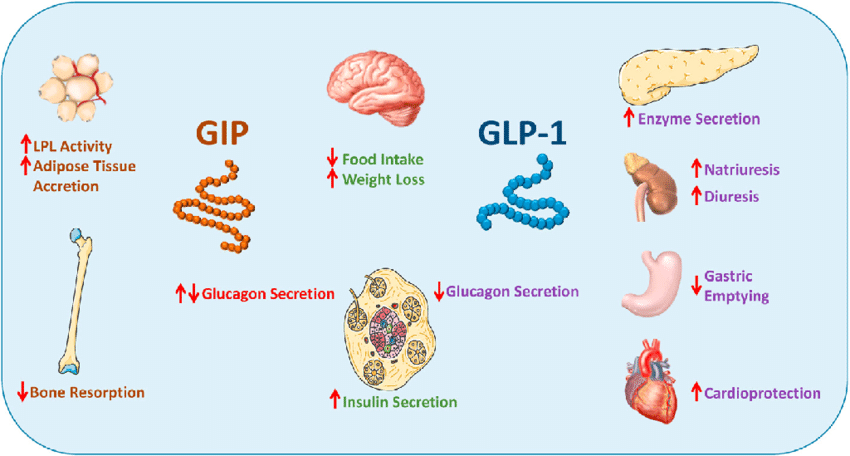
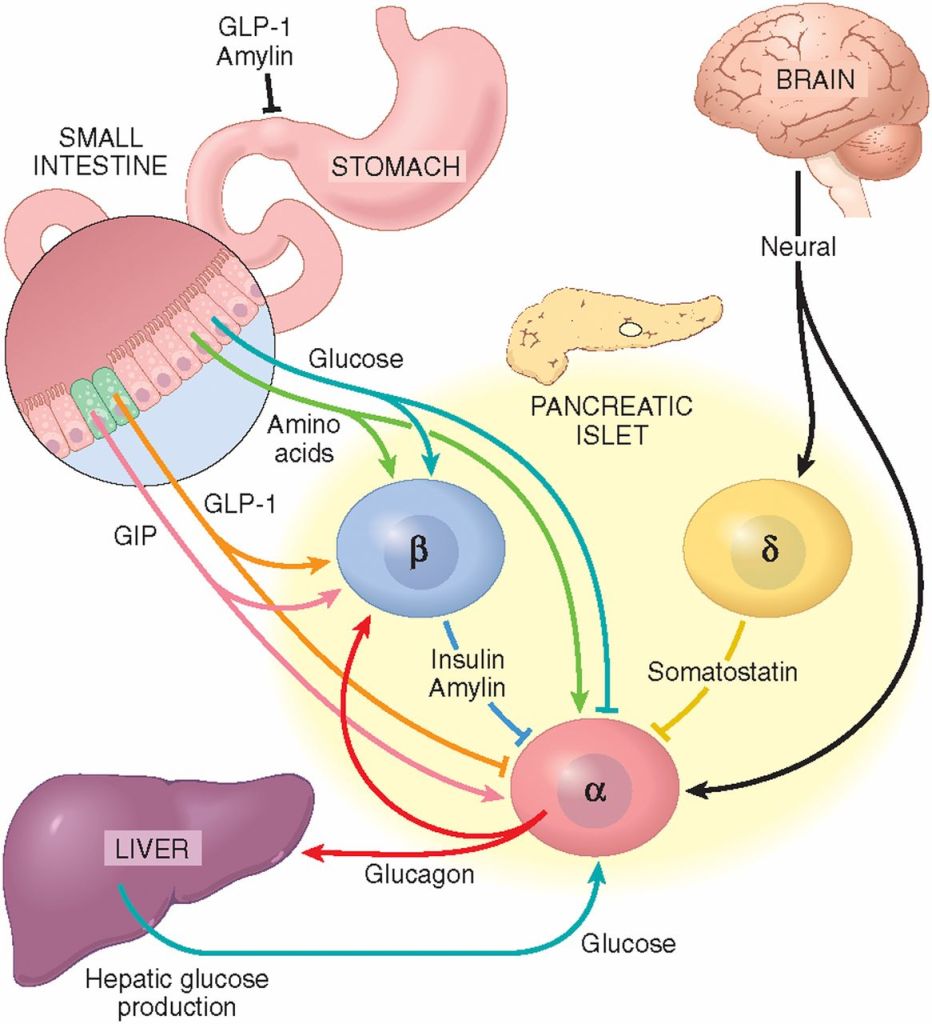
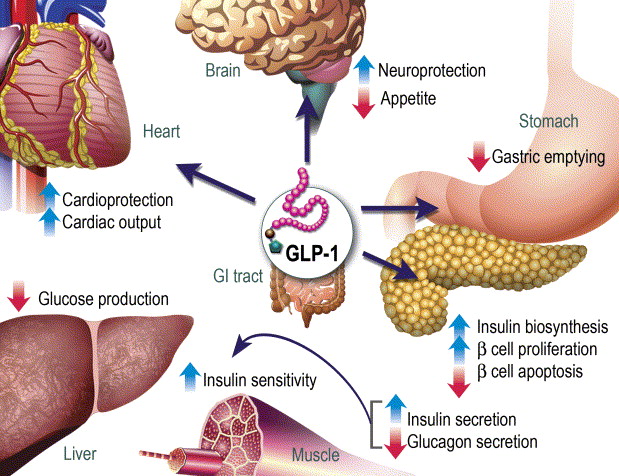
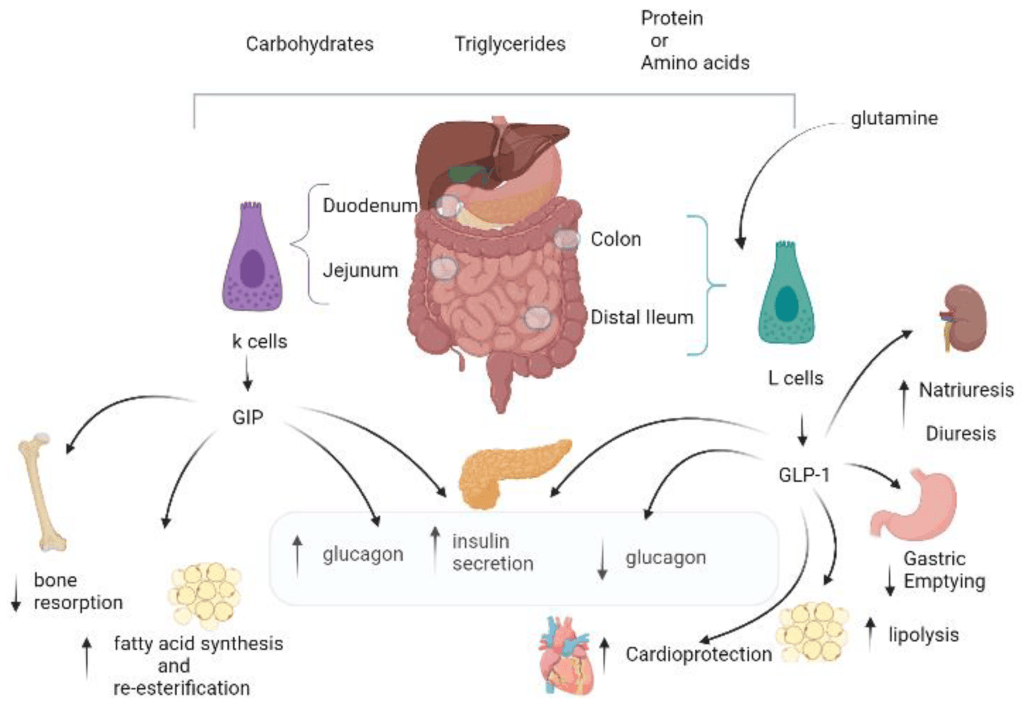
Gli ormoni incretinici GLP-1 e il GIP sono ormoni peptidici intestinali rilasciati in risposta all’ingestione di cibo.(3) L’effetto più importante del GLP-1 e del GIP è la loro capacità di potenziare la secrezione di Insulina indotta dal Glucosio da parte del pancreas – il cosiddetto effetto incretinico. Nei soggetti sani l’effetto incretinico rappresenta fino al 70% dell’Insulina secreta in risposta all’ingestione di Glucosio.(4) Il GLP-1 è un polipeptide di 30 aminoacidi sintetizzato dal proglucagone nelle cellule endocrine L distribuite principalmente nella mucosa della parte distale dell’intestino tenue e del colon. Il GIP è un polipeptide di 42 aminoacidi rilasciato dalle cellule endocrine K presenti nella mucosa del duodeno e della parte superiore del digiuno.(5) Mentre il GLP-1 viene rapidamente degradato (dall’enzima ubiquitario dipeptidil peptidasi-4 (DPP-4)) in circolo con un’emivita apparente di 1 – 1,5 minuti,(6) il GIP viene degradato più lentamente, con un’emivita per l’ormone intatto di 7 minuti.(7) L’ormone aumenta la secrezione di Insulina all’inizio del pasto, ma non ha alcuna attività insulinotropica a concentrazioni di glucosio inferiori (meno di 4mM), non favorendo così l’ipoglicemia. Il GLP-1 aumenta anche la biosintesi dell’Insulina e l’espressione genica della stessa. Inoltre, esercita azioni trofiche e protettive sulle beta-cellule(8) e inibisce fortemente la secrezione pancreatica di Glucagone in modo glucosio-dipendente.(9) Al contrario, è stato dimostrato che la GIP stimola la secrezione di Glucagone. Gli ormoni esercitano il loro effetto insulinotropico attraverso recettori accoppiati a proteine G sulle beta-cellule pancreatiche.(10) Oltre agli effetti sul pancreas endocrino, entrambi gli ormoni hanno diverse altre funzioni. I recettori del GLP-1 si trovano in varie regioni del cervello (11) e, una volta attivati, promuovono il senso di sazietà che, in combinazione con l’inibizione della motilità gastrointestinale indotta dal GLP-1 (mediata dal nervo vago (12) ), riduce l’assunzione di cibo e, consequenzialmente, il peso corporeo. I recettori del GLP-1 si trovano anche nel cuore (13) e la maggior parte dei dati suggerisce che il GLP-1 esercita effetti protettivi sul miocardio. È stato inoltre riscontrato che il GLP-1 riduce l’aumento postprandiale dei trigliceridi e abbassa la concentrazione di acidi grassi liberi nell’uomo.(14) Infine, studi su animali e sull’uomo indicano che il GLP-1 ha proprietà natriuretiche e diuretiche attraverso la modulazione dello scambio renale Na+/H+ (15) – un meccanismo che potrebbe servire a ridurre la pressione sanguigna. La GIP non sembra avere effetti fisiologici sul tratto gastrointestinale, sull’appetito o sull’assunzione di cibo, ma può avere un ruolo nel metabolismo dei lipidi (16) e delle ossa.(17)
Azioni del GLP-1 nei tessuti periferici. Il GLP-1 agisce direttamente sul pancreas endocrino, sul cuore, sullo stomaco e sul cervello, mentre le azioni su fegato e muscoli sono indirette.
Nei pazienti con diabete di tipo II l’effetto incretinico è gravemente ridotto.(18, 19) Questo tratto fisiopatologico gioca probabilmente un ruolo centrale nell’incapacità di questi pazienti di secernere una quantità di Insulina sufficiente a prevenire l’iperglicemia in seguito all’assunzione di glucosio per via orale.(20-30) L’attenuata secrezione postprandiale (21) e la diminuita potenza insulinotropica del GLP-1 (22), in combinazione con l’abolizione dell’effetto insulinotropico del GIP (23) , sembrano essere responsabili del ridotto effetto incretinico nei pazienti con diabete di tipo II. Poiché l’effetto insulinotropico del solo GLP-1 (e non del GIP) è conservato nei pazienti con diabete di tipo II, sono state sviluppate modalità di trattamento antidiabete basate sull’effetto di questo peptide. È interessante notare che l’infusione endovenosa (iv) di GLP-1 nativo è in grado di normalizzare la glicemia nei pazienti con diabete di tipo 2, (24) ma a causa della breve emivita del GLP-1, la somministrazione terapeutica del GLP-1 nativo non è praticabile. Pertanto, per sfruttare le azioni benefiche del GLP-1 nel diabete di tipo II, sono stati sviluppati agonisti stabili a lunga durata d’azione del recettore del GLP-1 (incretino-mimetici). Nella sezione seguente verranno descritti gli incretino-mimetici disponibili, le loro caratteristiche e applicazioni.
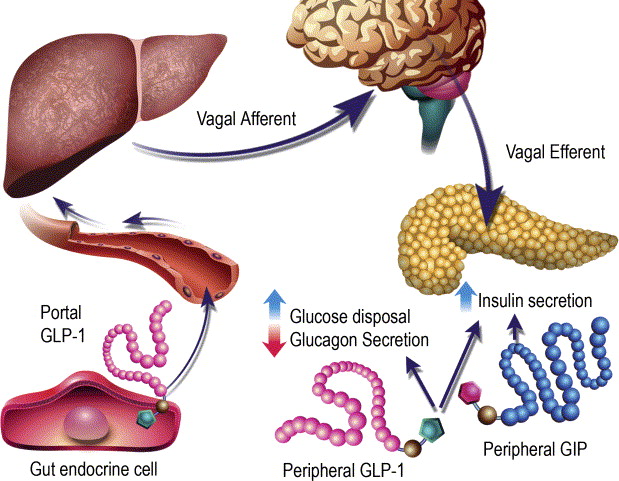
Ruoli contrastanti di GLP-1 e GIP sull’omeostasi del glucosio. Il GLP-1 secreto nella vena porta attiva un sensore del glucosio portale che segnala, tramite afferenze vagali, il sistema nervoso centrale e, a sua volta, le efferenze vagali aumentano la secrezione di Insulina. Sia il GLP-1 che il GIP attivano anche direttamente la secrezione di Insulina attraverso il legame con i loro recettori distinti sulle cellule β dell’isolotto.
Farmaci Incretino-mimetici:
Exenatide[approvato nel 2005/2012]
L’Exenatide, il primo di questa classe di farmaci, è stato introdotto sul mercato negli Stati Uniti nel 2005 e in Europa nel 2007 con il nome commerciale Byetta® (Amylin Pharmaceuticals/Eli Lilly).
L’Exenatide è un peptide di 39 aminoacidi; è una versione sintetica dell’exendin-4, un peptide presente nel veleno del “mostro di Gila”.
L’ Exenatide è stata isolata per la prima volta nel 1992 presso il Veterans Administration Medical Center di New York City.[25]
L’Exenatide si lega al recettore umano intatto del Peptide Glucagone-Simile-1 (GLP-1R) in modo simile al GLP-1; l’Exenatide ha un’omologia aminoacidica del 50% con il GLP-1 e ha un’emivita più lunga in vivo.[26]
Si ritiene che l’Exenatide faciliti il controllo del glucosio in almeno cinque modi:
L’Exenatide aumenta la risposta del Pancreas[27] (cioè aumenta la secrezione di Insulina) in risposta al consumo dei pasti; il risultato è il rilascio di una quantità di Insulina maggiore e più appropriata che aiuta a ridurre l’aumento della glicemia dovuto al consumo di cibo. Una volta che i livelli di glucosio nel sangue si avvicinano ai valori normali, la risposta del Pancreas alla produzione di Insulina si riduce; altri farmaci (come la rInsulina ) sono efficaci nell’abbassare la glicemia, ma possono “superare” il loro obiettivo e causare un abbassamento eccessivo della glicemia, provocando la pericolosa condizione di ipoglicemia.
L’Exenatide sopprime anche il rilascio di Glucagone da parte del Pancreas in risposta al pasto, impedendo al fegato di produrre eccessivamente glucosio quando non è necessario e prevenendo così l’iperglicemia (livelli elevati di glucosio nel sangue).
L’Exenatide contribuisce a rallentare lo svuotamento gastrico e quindi a ridurre la velocità di comparsa nel sangue del glucosio derivato dai pasti.
L’Exenatide ha un effetto sottile ma prolungato di riduzione dell’appetito e di promozione della sazietà attraverso i recettori ipotalamici (recettori diversi da quelli dell’Amilina). La maggior parte delle persone che utilizzano l’Exenatide perdono lentamente peso e, in genere, la perdita di peso maggiore è ottenuta dalle persone più in sovrappeso all’inizio della terapia con l’Exenatide. Gli studi clinici hanno dimostrato che l’effetto di riduzione del peso continua allo stesso ritmo per 2,25 anni di uso continuato. Se suddivisi in quartili di perdita di peso, il 25% più alto registra una sostanziale perdita di peso, mentre il 25% più basso non registra alcuna perdita o un lieve aumento di peso.
L’Exenatide riduce il contenuto di grasso nel fegato. L’accumulo di grasso nel fegato o la malattia del fegato grasso non alcolico (NAFLD) è fortemente correlata a diversi disturbi metabolici, in particolare a un basso livello di HDL e a Trigliceridi elevati, presenti nei pazienti con diabete di tipo II. È emerso che l’Exenatide ha ridotto il grasso epatico nei topi[28], nei ratti[29] e nell’uomo.[30]
I principali effetti collaterali del Exenatide (che condivide con tutte le molecole apopartenenti alla sua “famiglia”) sono nausea e vomito da lievi a moderati e transitori. L’incidenza dell’ipoglicemia associata al trattamento è bassa (31) – apparentemente dovuta agli effetti insulinotropi e glucagonostatici del GLP-1, dipendenti dal glucosio. Tuttavia, in combinazione con altri ipoglicemizzanti l’incidenza aumenta e dipende dalla dose di questi. Nella maggior parte degli studi con Exenatide gli episodi ipoglicemici minori sono definiti come glucosio plasmatico <3,3mM; negli studi LEAD sono definiti come glucosio plasmatico <3,1 mM. Negli studi che utilizzano Exenatide in combinazione con ipoglicemizzanti il rischio di episodi ipoglicemici minori è riportato tra il 15% e il 36%.(32) Nello studio Exenatide/Insulina Glargine l’1,5% dei pazienti ha sperimentato un’ipoglicemia grave.(33) Non c’è stata differenza tra i gruppi e l’incidenza è stata simile nei due gruppi.
Circa il 40% dei pazienti trattati con Exenatide in studi a lungo termine, controllati con placebo, ha sviluppato anticorpi contro il peptide durante le prime 30 settimane di trattamento.(32) L’esatto impatto degli anticorpi a lungo termine deve essere stabilito. L’Exenatide non è raccomandata durante la gravidanza o l’allattamento a causa della mancanza di dati sufficienti. L’Exenatide non deve essere utilizzata in pazienti con insufficienza renale, poiché viene eliminata principalmente nei reni attraverso la filtrazione glomerulare (31) e sono stati segnalati casi di insufficienza renale acuta. Nessun dato indica l’inibizione o l’induzione degli enzimi di metabolizzazione dei farmaci del citocromo P450. Dal 2005 la Food and Drug Administration (FDA) statunitense ha ricevuto 170 segnalazioni di pancreatite in pazienti trattati con Exenatide e ha ricevuto segnalazioni di pancreatite acuta, alcune delle quali erano casi gravi di pancreatite emorragica o necrotizzante in pazienti che assumevano Exenatide. Negli studi LEAD sono state osservate in totale (al 2010) 9 segnalazioni di pancreatite, nei pazienti trattati con Exanatide. E’ tutt’ora poco chiaro il nesso causale tra pancreatite ed Exenatide. Tuttavia, si raccomanda di interrompere il trattamento con incretino-mimetici in caso di sospetta pancreatite.(34) È stato suggerito che i risultati sul cancro nei roditori siano causati da un meccanismo non genotossico e specifico mediato dal recettore GLP-1 a cui i roditori sono particolarmente sensibili. La rilevanza per l’uomo è probabilmente insignificante dal punto di vista clinico, ma sono necessari ulteriori studi per chiarire i potenziali meccanismi alla base dello sviluppo del tumore delle cellule C nei pazienti trattati con analoghi del GLP-1 e le loro possibili implicazioni cliniche.(35)
Nel marzo 2013, la FDA ha pubblicato una Drug Safety Communication in cui annunciava l’avvio di indagini sui mimetici dell’incretina a causa dei risultati ottenuti da ricercatori accademici.[36] Poche settimane dopo, l’Agenzia europea per i medicinali ha avviato un’indagine simile sugli agonisti del GLP-1 e sugli inibitori della DPP-4.[37]
53 cause consolidate contro i produttori di “prodotti GLP-1/DPP-4” sono state archiviate nel 2015.[38]
Nel 2016 è stato pubblicato un lavoro che dimostra che è in grado di invertire l’alterata segnalazione del calcio nelle cellule epatiche steatotiche, che a sua volta potrebbe essere associata a un corretto controllo del glucosio.[39]
È in fase di valutazione per il trattamento del morbo di Parkinson.[40] Uno studio clinico di fase 3, iniziato nel gennaio 2020, ha avuto la sua data di completamento il 30 giugno 2024 (NCT04232969).[41]
L’Exenatide si presenta come soluzione parenterale destinata all’iniezione. Può essere somministrata nel sottocute dell’addome, del braccio o della coscia. Per migliorare la tollerabilità gastrointestinale, la terapia con Exenatide deve essere iniziata a 5mcg per dose somministrata due volte al giorno per almeno un mese. La dose di Exenatide può poi essere aumentata a 10mcg due volte al giorno.(31) La soddisfazione dei pazienti che hanno utilizzato Exenatide è stata valutata in un paio di studi. L’aggiunta di Exenatide a Metformina e altri ipoglicemizzanti ha determinato un miglioramento significativo della soddisfazione per il trattamento e della qualità di vita correlata alla salute dei pazienti dal basale a 26 settimane.(42) Il miglioramento è stato simile per i pazienti trattati con Insulina Glargine.
Liraglutide [approvato nel 2010]
La Liraglutide è un analogo acilato del GLP-1 umano e presenta il 97% di omologia di sequenza con il GLP-1 nativo. L’analogo è prodotto con la tecnologia del DNA ricombinante nel lievito.(43) Ha un effetto sul recettore del GLP-1 simile a quello descritto per l’Exenatide. Un elevato grado di legame con le proteine plasmatiche causa una minore suscettibilità al metabolismo da parte della DPP-4 e l’emivita dopo la somministrazione di Liraglutide è di circa 13 ore.(44) Questo profilo d’azione prolungato rende Liraglutide adatto alla somministrazione una volta al giorno. Non ci sono differenze clinicamente significative nella farmacocinetica di Liraglutide tra soggetti di sesso maschile e femminile, soggetti di razza diversa o soggetti anziani e giovani.(45)
La Liraglutide, venduta tra l’altro con i marchi Victoza e Saxenda, è un farmaco antidiabetico utilizzato per il trattamento del diabete di tipo II e dell’obesità cronica.[46][47] Si tratta di una terapia di seconda linea per il diabete dopo la terapia di prima linea con la Metformina.[46][48] Non sono chiari i suoi effetti sugli esiti di salute a lungo termine, come le malattie cardiache e l’aspettativa di vita.[46][49] Viene somministrata mediante iniezione sotto cutanea.[46]
La Liraglutide è stata approvata per uso medico nell’Unione Europea nel 2009 e negli Stati Uniti nel 2010.[50][51] Nel 2021 è stato il 166° farmaco più comunemente prescritto negli Stati Uniti, con oltre 3 milioni di prescrizioni.[52][53]
L’azione prolungata della Liraglutide si ottiene attaccando una molecola di acido grasso in una posizione della molecola GLP-1-(7-37), consentendole di auto-associarsi e di legarsi all’albumina nel tessuto sottocutaneo e nel flusso sanguigno. Il GLP-1 attivo viene quindi rilasciato dall’albumina a un ritmo lento e costante. Il legame con l’albumina determina inoltre una degradazione più lenta e un’eliminazione renale ridotta rispetto a quella del GLP-1-(7-37).[54]
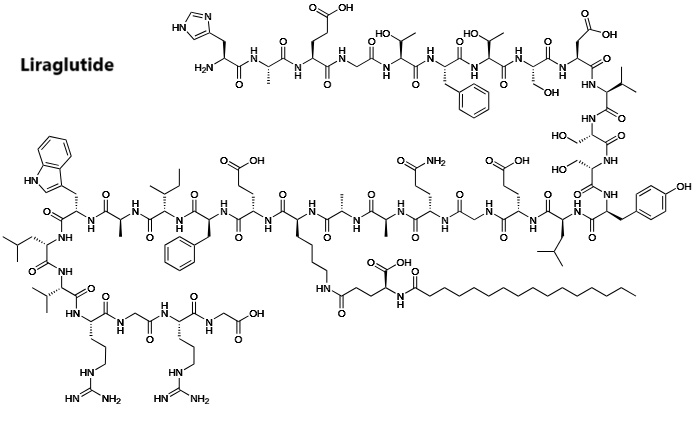
A) La struttura molecolare del GLP-1 umano. B) La struttura molecolare di Exenatide (il colore grigio indica le differenze di struttura rispetto al GLP-1 umano). C) La struttura molecolare della Liraglutide (il colore grigio indica le differenze di struttura rispetto al GLP-1 umano).
Come abbiamo visto, La Liraglutide è un agonista acilato del recettore del GLP-1, derivato dal GLP-1-(7-37) umano, una forma meno comune di GLP-1 endogeno.
Riduce l’iperglicemia correlata ai pasti (per 24 ore dopo la somministrazione) aumentando la secrezione di Insulina (solo) quando richiesto dall’aumento dei livelli di glucosio, ritardando lo svuotamento gastrico e sopprimendo la secrezione prandiale di Glucagone.[54][55]
Quindi, la Liraglutide provoca il rilascio di Insulina nelle cellule beta pancreatiche in presenza di una glicemia elevata. Questa secrezione di Insulina si attenua quando le concentrazioni di glucosio diminuiscono e si avvicinano all’euglicemia (livello normale di glucosio nel sangue). Diminuisce inoltre la secrezione di Glucagone in modo glucosio-dipendente e ritarda lo svuotamento gastrico. A differenza del GLP-1 endogeno, la Liraglutide è stabile contro la degradazione metabolica da parte delle peptidasi, con un’emivita plasmatica di 13 ore.[56][54]
Nei pazienti ad alto rischio cardiovascolare, è stato dimostrato che la Liraglutide riduce il rischio di morte per cause cardiovascolari, infarto miocardico non fatale o ictus non fatale. Le linee guida dell’ADA considerano attualmente Liraglutide una terapia farmacologica di prima linea per il diabete di tipo II (di solito insieme alla Metformina), in particolare per i pazienti con malattie cardiovascolari aterosclerotiche o obesità.[57] Una revisione Cochrane del 2011 ha dimostrato una riduzione dell’HbA1c dello 0,24% in più con Liraglutide. Del 24% in più con Liraglutide a 1,8 mg rispetto a Insulina Glargine, 0,33% in più rispetto a Exenatide 10mcg due volte al giorno, Sitagliptin e Rosiglitazone. In uno studio randomizzato e controllato (RCT) che ha confrontato Liraglutide, Insulina Glargine, Glimepiride e Sitagliptin (tutti aggiunti alla Metformina) con un follow-up di cinque anni, Insulina Glargine e Liraglutide sono risultate modestamente più efficaci nel raggiungimento e nel mantenimento dell’HbA1c target,[58] senza alcuna differenza negli esiti delle malattie microvascolari e cardiovascolari.[59]
La Liraglutide può anche essere utilizzata insieme alla dieta e all’esercizio fisico per la gestione cronica del peso negli adulti.[46] La Liraglutide ha portato a una perdita di peso maggiore rispetto ad alcuni precedenti analoghi del peptide glucagone-simile,[60] ma è meno efficace della dose standard di Semaglutide per la perdita di peso.[61][62]
In un recente studio pubblicato nel settembre 2024, Liraglutide ha aiutato i bambini di età compresa tra i 6 e i 12 anni a ridurre l’indice di massa corporea del 7,4% in uno studio di 56 settimane.[63] Se da un lato lo studio ha mostrato i potenziali benefici del farmaco, dall’altro solleva preoccupazioni riguardo all’uso di farmaci contro l’obesità in bambini così piccoli.[64] Novo Nordisk, l’azienda innovatrice che commercializza Liraglutide, ha chiesto alle autorità di regolamentazione statunitensi ed europee di estendere l’approvazione di Saxenda anche a questa fascia d’età più giovane, dato che attualmente è approvato solo per adolescenti e adulti.[65]
aUso di 1.8mg di Liraglutide.
Come per l’Exenatide, la Liraglutide ha un effetto significativo sul peso corporeo, come dimostrano i dati relativi a Liraglutide somministrata a 1,8mg/die. Liraglutide ha ridotto il peso corporeo medio o è stato neutro rispetto al placebo o ai comparatori attivi, in monoterapia (66) e in combinazione con uno (67) o due (68) agenti antidiabete orali. Lo studio LEAD 662 ha esaminato il profilo lipidico con Exenatide e Liraglutide. Sono state osservate riduzioni significative maggiori dei trigliceridi (-0,4 vs -0,2 mM) e degli acidi grassi liberi (-0,17 vs -0,10 mM) nel gruppo Liraglutide. Entrambi i composti hanno causato una riduzione significativa della pressione arteriosa (pressione sistolica -2,2 mmHg e pressione diastolica -1,5 mmHg) senza differenze significative tra i due composti.
Tra gli effetti collaterali si annoverano ipoglicemia, nausea, vertigini, dolore addominale e dolore nel sito di iniezione.[46] Gli effetti collaterali gastrointestinali tendono a essere più forti all’inizio del periodo di trattamento e si attenuano con il tempo.[60] Altri effetti collaterali gravi possono includere angioedema, pancreatite, malattie della cistifellea e problemi renali. L’uso in gravidanza e durante l’allattamento non è sicuro.[46] Una black box warning avverte che nei ratti trattati con Liraglutide sono stati osservati tumori midollari della tiroide, ma è “Sconosciuto se Liraglutide causi tumori delle cellule C della tiroide, incluso il carcinoma midollare della tiroide (MTC), nell’uomo, poiché la rilevanza per l’uomo di tali tumori nei roditori non è stata determinata.”[46]
A proposito del MTC, a esposizioni otto volte superiori a quelle utilizzate nell’uomo, la Liraglutide ha causato un aumento statisticamente significativo dei tumori alla tiroide nei ratti. La rilevanza clinica di questi risultati è sconosciuta.[69] Negli studi clinici, il tasso di tumori alla tiroide nei pazienti trattati con Liraglutide è stato di 1,3 per 1000 anni-paziente (4 persone) rispetto a 1,0 per 1000 pazienti (1 persona) nei gruppi di confronto. L’unica persona nel gruppo di confronto e quattro delle cinque persone nel gruppo Liraglutide avevano marcatori sierici (calcitonina elevata) suggestivi di una malattia preesistente al basale.[69]
L’FDA ha dichiarato che la calcitonina sierica, un biomarcatore del carcinoma midollare della tiroide, era leggermente aumentata nei pazienti con Liraglutide, ma ancora nei limiti della norma, e che era necessario un monitoraggio continuo per 15 anni in un registro dei tumori.[70]
Un altro effetto collaterale preoccupante è rappresentato dalla possibilità (sebbene rara) di sviluppare pancreatite.
Nel 2013, un gruppo della Johns Hopkins ha riportato un’associazione con apparenza statisticamente significativa tra l’ospedalizzazione per pancreatite acuta e un precedente trattamento con derivati del GLP-1 (come la precedentemente vista Exenatide) e inibitori della DPP-4 (come il Sitagliptin).[71] In risposta, la FDA degli Stati Uniti e l’Agenzia Europea per i Medicinali hanno condotto una revisione di tutti i dati disponibili in merito alla possibile connessione tra i mimetici dell’Incretina e la pancreatite o il cancro al pancreas. In una lettera congiunta del 2014 al New England Journal of Medicine, le agenzie hanno concluso che “Un’analisi congiunta dei dati di 14.611 pazienti con diabete di tipo II provenienti da 25 studi clinici nel database di sitagliptin non ha fornito alcuna prova convincente di un aumento del rischio di pancreatite o di cancro al pancreas” e “Entrambe le agenzie concordano sul fatto che le affermazioni relative a un’associazione causale tra i farmaci a base di Incretine e la pancreatite o il cancro al pancreas, espresse di recente nella letteratura scientifica e nei media, non sono coerenti con i dati attuali”. L’FDA e l’EMA non hanno ancora raggiunto una conclusione definitiva su tale relazione causale. Sebbene la totalità dei dati esaminati fornisca rassicurazioni, la pancreatite continuerà a essere considerata un rischio associato a questi farmaci finché non saranno disponibili ulteriori dati; entrambe le agenzie continuano a indagare su questo segnale di sicurezza”[72].
Albiglutide [approvato nel 2014]
L’Albiglutide (nome commerciale Eperzan in Europa e Tanzeum negli Stati Uniti) è un farmaco agonista del GLP-1 commercializzato da GlaxoSmithKline (GSK) per il trattamento del diabete di tipo II.
L’Albiglutide è un peptide composto da 645 aminoacidi proteinogenici con 17 ponti disolfuro. Gli aminoacidi 1-30 e 31-60 costituiscono due copie di GLP-1 umano modificato, in cui l’alanina in posizione 2 è stata scambiata con una glicina per migliorare la resistenza alla DPP-4.[73] La sequenza rimanente è costituita da albumina umana.
Viene bioingegnerizzata nel lievito Saccharomyces cerevisiae utilizzando la tecnologia del DNA ricombinante.[74]
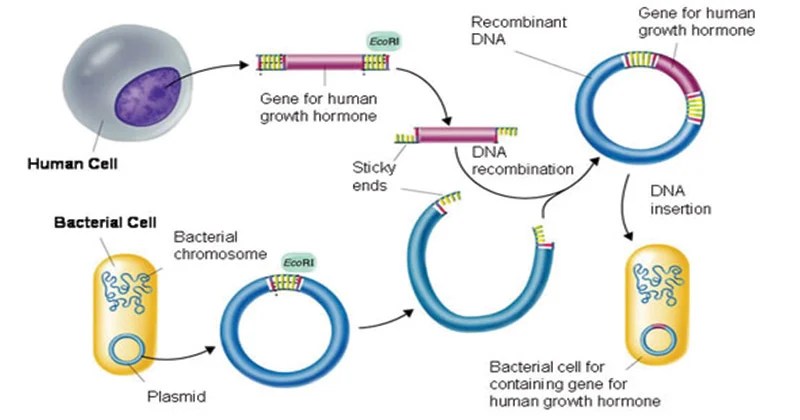
Fasi applicative della tecnologia del DNA ricombinante.
Il farmaco è stato brevettato dalla Human Genome Sciences e sviluppato in collaborazione con GSK.[75]
La GSK ha presentato domanda di approvazione alla FDA statunitense il 14 gennaio 2013 e all’Agenzia europea per i medicinali (EMA) il 7 marzo 2013. Nel marzo 2014, GSK ha ricevuto dalla Commissione Europea l’approvazione per la commercializzazione di Albiglutide con il nome di Eperzan.[76] Nell’aprile 2014, la FDA statunitense ha approvato Albiglutide con il nome di Tanzeum.[77]
Nell’agosto 2017, GSK annunciò l’intenzione di ritirare il farmaco dal mercato mondiale entro luglio 2018 per motivi economici.[78]
L’Albiglutide, come gli altri incretino-mimetici, è stato utilizzato per il trattamento del diabete di tipo II negli adulti. Può essere utilizzato da solo (se la terapia con Metformina è inefficace o non tollerata) o in combinazione con altri farmaci antidiabetici, comprese le forme di Insulina.[74]
Secondo un’analisi del 2015, l’Albiglutide è meno efficace di altri agonisti del GLP-1 per la riduzione dell’HbA1c e la perdita di peso. Sembra inoltre avere meno effetti collaterali rispetto alla maggior parte degli altri farmaci di questa classe, ad eccezione delle reazioni nel sito di iniezione che sono più comuni con Albiglutide rispetto, ad esempio, a Liraglutide.[79]
Dopo l’iniezione sottocutanea, l’Albiglutide raggiunge le massime concentrazioni ematiche dopo tre-cinque giorni. Le concentrazioni allo stato stazionario vengono raggiunte dopo tre-cinque settimane. Essendo resistente alla dipeptidil peptidasi-4 (DPP-4),[73] l’enzima che scompone il GLP-1, l’Albiglutide ha un’emivita biologica di cinque (da quattro a sette) giorni, notevolmente più lunga rispetto agli analoghi del GLP-1 più vecchi, l’Exenatide e la Liraglutide. [80][81] Ciò consente una somministrazione una volta alla settimana,[74] a differenza della Liraglutide ma come la forma a rilascio prolungato dell’Exenatide.
L’Albiglutide agisce come agonista del recettore GLP-1, il che lo rende un tipo di incretino-mimetico. Questo provoca un aumento della secrezione di insulina, soprattutto in presenza di glucosio elevato nel sangue, e rallenta anche lo svuotamento gastrico.[74]
La differenza nel meccanismo d’azione del Albiglutide con gli altri agonisti del recettore del GLP-1, dipende dalla sua struttura la quale rende difficile l’attraversamento della barriera emato-encefalica. Ciò significa che non influisce sul sistema nervoso centrale come altri agonisti del recettore del GLP-1 e potrebbe essere responsabile della limitata perdita di peso osservata con questo farmaco.[81]
Dulaglutide[approvato nel 2014]
La Dulaglutide, venduta tra l’altro con il nome commerciale Trulicity,[8] è un farmaco utilizzato per il trattamento del diabete di tipo II in combinazione con la dieta e l’esercizio fisico.[83][84] È inoltre approvato negli Stati Uniti per la riduzione degli eventi cardiovascolari avversi maggiori negli adulti con diabete di tipo II che presentano una malattia cardiovascolare conclamata o molteplici fattori di rischio cardiovascolare.[85]
Come per gli altri incretino-mimetici visti in precedenza, la Dulaglutide si lega ai recettori del GLP-1, rallentando lo svuotamento gastrico e aumentando la secrezione di Insulina da parte delle cellule β pancreatiche. Contemporaneamente, il peptide riduce l’elevata secrezione di Glucagone inibendo le cellule α del pancreas, poiché è noto che il Glucagone è elevato in modo inappropriato nei pazienti diabetici.
Più precisamente, la Dulaglutide è un agonista del recettore del GLP-1 costituito da GLP-1(7-37) legato covalentemente a un frammento Fc di IgG4 umana.
La sicurezza e l’efficacia della Dulaglutide sono state valutate in sei studi clinici in cui 3.342 soggetti con diabete di tipo II hanno ricevuto Dulaglutide. I soggetti che hanno ricevuto Dulaglutide hanno registrato un miglioramento del controllo glicemico, osservato con riduzioni del livello di HbA1c.[86]
La Food and Drug Administration (FDA) statunitense ha approvato la Dulaglutide con una strategia di valutazione e mitigazione dei rischi (REMS),[86] e ha concesso l’approvazione di Trulicity a Eli Lilly and Company.[86] La REMS consiste in una serie di misure che Eli Lilly adotterà per sensibilizzare i medici sul rischio di pancreatite e sul potenziale rischio di carcinoma midollare della tiroide associato al farmaco.[87]
Nel 2020, la FDA ha approvato due dosi più elevate del farmaco, 3,0mg e 4,5mg, sulla base dei risultati dello studio AWARD-11 che hanno dimostrato una migliore riduzione del glucosio e benefici sul peso.[88]
Il peptide è indicato per gli adulti con diabete di tipo II come aggiunta alla dieta e all’esercizio fisico per migliorare il controllo glicemico. La Dulaglutide non è indicato nel trattamento di soggetti con diabete di tipo I o di pazienti con chetoacidosi diabetica perché questi problemi sono dovuti all’incapacità delle isole pancreatiche di produrre Insulina e una delle azioni della Dulaglutide è quella di stimolare le isole funzionanti a produrre più Insulina. La Dulaglutide può essere utilizzata da solo o in combinazione con altri farmaci per il diabete di tipo II, in particolare Metformina, Sulfoniluree, Tiazolidinedioni e Insulina da assumere contemporaneamente ai pasti.[89]
Il programma di sperimentazione clinica di fase 3 del farmaco ha dimostrato riduzioni dell’emoglobina A1c di circa l’1% con le dosi di 0,75mg e 1,5mg del farmaco, insieme a una perdita di peso media di circa 5Kg. Le dosi più elevate da 3,0mg e 4,5mg, approvate nel 2020, hanno dimostrato riduzioni dell’emoglobina A1c più vicine all’1,5% e una perdita di peso leggermente superiore.[90]
DPP-4
Una meta-analisi del 2017 non ha supportato l’ipotesi che il trattamento con agonisti del GLP-1 o inibitori della DPP-4 aumenti la mortalità per tutte le cause nei diabetici di tipo II.[91]
La Dulaglutide viene assorbita lentamente dopo l’iniezione sottocutanea. In uno studio farmacocinetico condotto su 20 adulti sani, la Cmax si è verificata entro 24-48 ore dalla somministrazione. La biodisponibilità assoluta media di Dulaglutide dopo iniezioni sottocutanee di dosi singole da 0,75mg e 1,5mg è stata rispettivamente del 65% e del 47%. L’emivita media della Dulaglutide somministrato a varie dosi è stata di circa 3,75 giorni (89,9 ore). Questa emivita prolungata consente la somministrazione una volta alla settimana. Le informazioni di prescrizione indicano un’emivita di circa 5 giorni.
Gli effetti collaterali più comuni includono disturbi gastrointestinali, come dispepsia, inappetenza, nausea, vomito, dolore addominale, diarrea.[92] Alcuni pazienti possono manifestare reazioni avverse gravi: pancreatite acuta (i sintomi includono dolore addominale persistente e grave, che talvolta si irradia alla schiena ed è accompagnato da vomito), ipoglicemia, insufficienza renale (che talvolta può richiedere l’emodialisi). Il rischio di ipoglicemia aumenta se il farmaco è usato in combinazione con Sulfoniluree o Insulina.[93][94] Esiste anche un rischio potenziale di carcinoma midollare della tiroide associato all’uso del farmaco.[87]
Lixisenatide [approvato nel 2016]
La Lixisenatide (nome commerciale Lyxumia nell’Unione Europea e Adlyxin negli Stati Uniti e prodotto da Sanofi) è un agonista del recettore GLP-1 iniettabile una volta al giorno per il trattamento del diabete di tipo II.
È stato sintetizzato dalla danese Zealand Pharma A/S;[95] nel 2003 Zealand lo ha concesso in licenza a Sanofi, che ha sviluppato il farmaco.[96] La Lixisenatide è stata approvata dalla Commissione europea nel febbraio 2013.
La Lixisenatide è un peptide composto da 44 aminoacidi, con un gruppo amidico sul suo terminale C.[97]
E’ stata descritta come “des-38-prolina-exendin-4 (Heloderma suspectum)-(1-39)-peptidilpenta-L-lisil-L-lisinamide”, ovvero è derivata dai primi 39 aminoacidi della sequenza del peptide exendin-4, isolato dal veleno del “mostro di Gila”, omettendo la Prolina in posizione 38 e aggiungendo sei residui di Lisina. La sua sequenza completa è:
La Lixisenatide, appartenendo alla classe dei farmaci agonisti del GLP-1, come per i precedentemente trattati composti agisce rallentando lo svuotamento gastrico e aumentando la secrezione di Insulina da parte delle cellule β pancreatiche.
I risultati di una ricerca condotta da McClean PL et al. hanno dimostrato che la Liraglutide e la Lixisenatide sono promettenti come potenziali trattamenti farmacologici della malattia di Alzheimer AD. La Lixisenatide è risultata ugualmente efficace a una dose inferiore rispetto alla Liraglutide in alcuni dei parametri misurati dopo dieci settimane di iniezioni intraperitoneali giornaliere di Liraglutide (2,5 o 25 nmol/kg) o Lixisenatide (1 o 10 nmol/kg) o soluzione fisiologica in topi APP/PS1 a un’età in cui le placche amiloidi si erano già formate. Analizzando la plasticità sinaptica nell’ippocampo, l’LTP è stato fortemente aumentato nei topi APP/PS1 da entrambi i farmaci, con maggiore efficacia con la Lixisenatide. La riduzione del numero di sinapsi osservata nei topi APP/PS1 è stata evitata dai due farmaci. Il carico di placche amiloidi e il carico di placche Congo rosso positivo a nucleo denso nella corteccia sono stati ridotti da entrambi i farmaci a tutte le dosi. Anche la risposta infiammatoria cronica (attivazione microgliale) è stata ridotta da tutti i trattamenti.[98]
Cai HY et al. hanno dimostrato in uno studio che la lixisenatide è in grado di ridurre le placche amiloidi, i grovigli neurofibrillari e la neuroinfiammazione negli ippocampi di topi femmina APP/PS1/tau di 12 mesi; l’attivazione della via di segnalazione PKA-CREB e l’inibizione della p38-MAPK potrebbero essere i meccanismi importanti nella funzione neuroprotettiva della lixisenatide. Pertanto, la lixisenatide potrebbe avere il potenziale per essere sviluppata come nuova terapia per l’AD. [99] Liu Wet al hanno trovato risultati interessanti confrontando exendin-4 (10 nmol/kg), liraglutide (25 nmol/kg) e lixisenatide (10 nmol/kg): è emerso che exendin-4 non ha mostrato effetti protettivi alla dose scelta, mentre sia liraglutide che lixisenatide hanno mostrato effetti nel prevenire la compromissione motoria indotta da MPTP (Rotarod, locomozione in campo aperto, test di catalessi), la riduzione dei livelli di tirosina idrossilasi (TH) (sintesi di dopamina) nella substantia nigra e nei gangli della base, una riduzione della molecola di segnalazione pro-apoptotica BAX e un aumento della molecola di segnalazione anti-apoptotica B-cell lymphoma-2. I risultati precedenti dimostrano che sia la liraglutide che la lixisenatide sono superiori all’exendin-4 ed entrambi i farmaci sono promettenti come nuovo trattamento della malattia di Parkinson.[100]
Un altro studio condotto da Kerry Hunter et al. ha analizzato gli agonisti del recettore GLP-1 liraglutide e lixisenatide. Sono state valutate le cinetiche di attraversamento della barriera ematoencefalica (BBB), l’attivazione del GLP-1R attraverso la misurazione dei livelli di cAMP e gli effetti fisiologici nel cervello sulla proliferazione delle cellule staminali neuronali e sulla neurogenesi. Entrambi i farmaci sono stati in grado di attraversare la BBB. La lixisenatide ha attraversato la BBB a tutte le dosi testate (2,5, 25 o 250 nmol/kg ip.) quando misurate 30 minuti dopo l’iniezione e a 2,5-25 nmol/kg ip. 3 ore dopo l’iniezione. La lixisenatide ha anche aumentato la neurogenesi nel cervello. La liraglutide ha attraversato la BBB a 25 e 250 nmol/kg ip. ma nessun aumento è stato rilevato a 2,5 nmol/kg ip. 30 minuti dopo l’iniezione, e a 250 nmol/kg ip. a 3 ore dopo l’iniezione. Liraglutide e lixisenatide hanno aumentato i livelli di cAMP nel cervello, con lixisenatide più efficace. I risultati precedenti suggeriscono che questi nuovi analoghi dell’incretina attraversano la BBB mostrando attività fisiologica e neurogenesi nel cervello, il che li rende buoni candidati per essere utilizzati come trattamento delle malattie neurodegenerative.[101]
Anche la Lixisenatide è utilizzata come coadiuvante della dieta e dell’esercizio fisico per il trattamento del diabete di tipo II.[97] Nell’Unione Europea il suo uso è limitato all’integrazione della terapia Insulinica.[102][103] Al 2017 non è chiaro se influisca sul rischio di morte di una persona.[104]
Viene fornito in un autoiniettore contenente quattordici dosi e viene iniettato per via sottocutanea.[97]
La Lixisenatide non deve essere utilizzata da persone che hanno problemi di svuotamento gastrico.[97] La Lixisenatide ritarda lo svuotamento gastrico, il che può modificare la velocità con cui altri farmaci assunti oralmente esplicano la loro efficacia.[97]
Dopo la somministrazione sottocutanea nell’uomo, la Lixisenatide mostra una farmacocinetica lineare e un’emivita di eliminazione dipendente dall’assorbimento di 2-3 ore.
La dose iniziale di Lixisenatide è di 10mcg una volta al giorno, per 14 giorni. La dose di mantenimento è successivamente di 20mcg una volta al giorno nell’ora che precede il primo pasto della giornata o il pasto serale.
In circa lo 0,1% dei casi le persone hanno avuto reazioni anafilattiche alla lixisenatide e in circa lo 0,2% dei casi il farmaco ha causato pancreatite.[97] L’uso con insulina o sulfonilurea può causare ipoglicemia.[97] In alcuni casi, persone senza malattie renali hanno avuto lesioni renali acute e in alcune persone con malattie renali esistenti la condizione è peggiorata. Poiché la Lixisenatide è un peptide, le persone possono sviluppare una risposta immunitaria nei suoi confronti che finirà per rendere il farmaco inefficace; le persone che hanno sviluppato anticorpi contro la Lixisenatide tendono ad avere una maggiore infiammazione nel sito di iniezione.[97]
Almeno il 5% delle persone ha avuto nausea, vomito, diarrea, mal di testa o vertigini dopo l’assunzione di Lixisenatide.[97]
Semaglutide [approvata nel 2017]
La Semaglutide è chimicamente simile al GLP-1 umano.[105-41] Mancano i primi sei aminoacidi del GLP-1.[105] Le sostituzioni sono effettuate nelle posizioni 8 e 34 del GLP-1 (posizioni 2 e 28 della Semaglutide), dove l’Alanina e la Lisina sono sostituite rispettivamente dall’acido 2-aminoisobutirrico e dall’Arginina. La sostituzione dell’Alanina impedisce la degradazione chimica da parte della dipeptidil peptidasi-4.[106] La Lisina in posizione 26 del GLP-1 (posizione 20 del Semaglutide) ha una lunga catena attaccata, che termina con una catena di 17 atomi di carbonio e un gruppo carbossilico.[106] Ciò aumenta il legame del farmaco con le proteine trasportatrici nel sangue (albumina), consentendo una più lunga presenza nella circolazione sanguigna.[106]
L’emivita del Semaglutide nel sangue è di circa sette giorni (165-184 ore).
Come per gli altri incretino-mimetici, la Semaglutide è un agonista del recettore del GLP -1.[107][108][109] Il farmaco riduce i livelli di glucosio nel sangue. Sembra inoltre che aumenti la crescita delle cellule β pancreatiche, responsabili della produzione e del rilascio di Insulina.[110][111] Inoltre, inibisce la produzione di Glucagone, l’ormone che aumenta la glicogenolisi (rilascio dei carboidrati immagazzinati dal fegato) e la Gluconeogenesi (sintesi di nuovo glucosio). Riduce l’assunzione di cibo abbassando l’appetito e rallentando la digestione nello stomaco e suo svuotamento,[112] contribuendo a ridurre il peso corporeo.[113][114]
Effetti di svuotamento gastrico degli agonisti del recettore del glucagone peptide-1 ad azione breve rispetto a quelli ad azione prolungata (GLP-1RA). (A) I GLP-1RA a breve durata d’azione sopprimono lo svuotamento gastrico, prolungando la presenza di cibo nello stomaco e nella parte superiore dell’intestino tenue; il ridotto flusso transpilorico provoca un ritardo nell’assorbimento intestinale del glucosio e una diminuzione della secrezione insulinica postprandiale. I GLP-1RA a breve durata d’azione possono anche sopprimere direttamente la secrezione di glucagone. (B) I GLP-1RA a lunga durata d’azione non influenzano significativamente la motilità gastrica, a causa della tachifilassi. Invece, i GLP-1RA ad azione prolungata esercitano maggiormente il loro effetto attraverso il pancreas, aumentando la secrezione di insulina e inibendo la secrezione di glucagone attraverso il rilascio paracrino di somatostatina. Agendo sul sistema nervoso centrale, sia i GLP-1RA a più breve (A) che a più lunga durata d’azione (B) aumentano la sazietà e possono anche indurre la nausea. Adattato da Meier. Adattato su autorizzazione di Macmillan Publishers Ltd: Nature Reviews Endocrinology 2012;8(12):728-42, copyright 2012.
Nel giugno 2008 è stato avviato uno studio clinico di fase II al fine di esaminare la Semaglutide come terapia per il diabete da somministrare una volta alla settimana, come alternativa ad azione prolungata alla Liraglutide .[115][116] Gli studi clinici sono iniziati nel gennaio 2016 e si sono conclusi nel maggio 2017.[117][118]
Nel giugno 2021, una versione iniettabile a dosaggio più elevato, venduta con il marchio Wegovy, è stata approvata dalla Food and Drug Administration (FDA) statunitense come farmaco anti-obesità per la gestione del peso a lungo termine negli adulti.[119-15] Nel novembre 2021, il Comitato per i Medicinali per Uso Umano (CHMP) dell’Agenzia Europea per i Medicinali (EMA) ha raccomandato di concedere a Novo Nordisk A/S l’autorizzazione all’immissione in commercio di Wegovy[120]. Nel gennaio 2022, Wegovy è stato approvato per uso medico nell’Unione Europea.[121]
Nel gennaio 2023, l’etichetta di Rybelsus è stata aggiornata per indicare che può essere utilizzato come trattamento di prima linea per gli adulti con diabete di tipo 2.[122]
Nel marzo 2021, in uno studio di fase III randomizzato, in doppio cieco, 1.961 adulti con un indice di massa corporea pari o superiore a 30 sono stati assegnati, in un rapporto 2:1, a un trattamento con Semaglutide sottocutaneo una volta alla settimana o placebo, più un intervento sullo stile di vita. Gli studi si sono svolti in 129 siti in 16 Paesi di Asia, Europa, Nord America e Sud America. La variazione percentuale media del peso corporeo alla settimana 68 è stata di -14,9% nel gruppo Semaglutide contro -2,4% con placebo, per una differenza di trattamento stimata di -12,4 punti percentuali (95% CI, da -13,4 a -11,5).[123][124][125][126]
Una revisione dei trattamenti anti-obesità del 2022 ha rilevato che il Semaglutide e la Tirzepatide (che ha un meccanismo d’azione sovrapponibile) erano più promettenti dei precedenti farmaci anti-obesità, anche se meno efficaci della chirurgia bariatrica.[127]
Nel marzo 2023, un funzionario di Novo Nordisk ha dichiarato che i pazienti che utilizzano la Semaglutide per perdere peso possono riacquistare il peso originario entro 5 anni dall’interruzione del trattamento.[128]
Nel marzo 2024, l’FDA ha esteso l’indicazione di Semaglutide (Wegovy) per ridurre il rischio di morte cardiovascolare, infarto e ictus in adulti con malattie cardiovascolari e obesità o sovrappeso. L’efficacia e la sicurezza di questa nuova indicazione sono state studiate in uno studio multinazionale, multicentrico, in doppio cieco, controllato con placebo, che ha assegnato in modo casuale oltre 17.600 partecipanti a ricevere Semaglutide (Wegovy) o placebo.[129] I partecipanti di entrambi i gruppi hanno ricevuto anche un trattamento medico standard (ad es, Semaglutide (Wegovy) ha ridotto significativamente il rischio di eventi cardiovascolari avversi maggiori (morte cardiovascolare, infarto e ictus), che si sono verificati nel 6,5% dei partecipanti che hanno ricevuto Semaglutide (Wegovy) rispetto all’8% dei partecipanti che hanno ricevuto placebo.[129]
Una meta-analisi del 2014 ha rilevato che la Semaglutide può essere efficace nell’abbassare gli enzimi epatici (transaminite) e nel migliorare alcune caratteristiche radiologicamente osservate della malattia epatica steatotica associata a disfunzione metabolica.[130]
Nel luglio 2023, l’Agenzia islandese per i medicinali ha segnalato due casi di pensieri suicidi e un caso di autolesionismo tra i consumatori del farmaco, inducendo a valutare la sicurezza di Ozempic,[131] Wegovy, Saxenda e altri farmaci simili.[132] Nel gennaio 2024, una revisione preliminare condotta dalla FDA ha confermato che non sono state trovate prove che suggeriscano che il farmaco causi pensieri o azioni suicide.[133][134]
La Semaglutide ha dimostrato di poter ridurre l’interesse per il consumo di alcol tra gli utilizzatori. Gli scienziati ipotizzano che il Semaglutide possa influenzare le regioni cerebrali coinvolte nella dipendenza e nella regolazione dell’appetito, sebbene i meccanismi esatti siano ancora in fase di studio. La ricerca sugli animali ha indicato che farmaci simili alla Semaglutide possono ridurre l’assunzione di alcolici.[135]
La Semaglutide e farmaci simili, come la Dulaglutide e la Liraglutide, sono stati utilizzati per trattare il disturbo da alimentazione incontrollata (BED), in quanto possono minimizzare i pensieri ossessivi sul cibo e gli impulsi ad abbuffarsi.[136][137] Alcuni utilizzatori di questi farmaci hanno riferito di aver ridotto in modo significativo quello che è colloquialmente noto come “food noise” (pensieri costanti e inarrestabili di mangiare nonostante non si abbia fisicamente fame), che può essere un fattore di BED.[138][139]
Attualmente, la Semaglutide indicata come coadiuvante della dieta e dell’esercizio fisico per migliorare il controllo glicemico negli adulti con diabete di tipo II.[140][141]
La formulazione a dosi più elevate di Semaglutide è indicata come coadiuvante della dieta e dell’esercizio fisico per la gestione del peso a lungo termine negli adulti con obesità (indice di massa corporea (IMC) iniziale ≥ 30 kg/m2) o in sovrappeso (IMC iniziale ≥ 27 kg/m2) e con almeno una comorbidità correlata al peso.[142]
Nel marzo 2024, la Food and Drug Administration (FDA) statunitense ha ampliato l’indicazione di Semaglutide (Wegovy), in combinazione con una dieta a ridotto contenuto calorico e un aumento dell’attività fisica, per ridurre il rischio di morte cardiovascolare, infarto e ictus in adulti obesi o in sovrappeso con malattie cardiovascolari.[143]
La dose iniziale è di 0,25mg di Semaglutide una volta alla settimana. Dopo 4 settimane, la dose deve essere aumentata a 0,5 mg una volta alla settimana. Dopo almeno 4 settimane con una dose da 0,5 mg una volta alla settimana, la dose può essere aumentata a 1 mg una volta alla settimana per migliorare ulteriormente il controllo glicemico. Dopo almeno 4 settimane con una dose da 1 mg una volta alla settimana, la dose può essere aumentata a 2 mg una volta alla settimana per migliorare ulteriormente il controllo glicemico.
Semaglutide 0,25mg non è una dose di mantenimento. Non sono raccomandate dosi superiori a 2 mg alla settimana.
Quando Ozempic viene aggiunto alla terapia in atto a base di Metformina e/o Tiazolidinedione o dell’ inibitore del cotrasportatore sodio-glucosio (SGLT2), la dose di Metformina e/o Tiazolidinedione o dell’inibitore SGLT2 può essere mantenuta senza variazioni.
Quando Ozempic viene aggiunto alla terapia in atto con Sulfanilurea o con un’insulina, è necessario considerare una riduzione della dose di Sulfanilurea o di insulina per ridurre il rischio di ipoglicemia (vedere paragrafi 4.4 e 4.8).
Non è necessario automonitorare la glicemia per aggiustare la dose di Ozempic. L’auto-monitoraggio della glicemia è necessario per correggere la dose di Sulfanilurea e insulina, in particolare quando si inizia Ozempic e si riduce l’insulina. Si raccomanda un approccio graduale alla riduzione dell’insulina.
Similmente agli altri incretino-mimetici, possibili effetti avversi con l’uso di questo peptide includono nausea, diarrea, vomito, costipazione, dolore addominale, cefalea, affaticamento, indigestione/bruciore di stomaco, vertigini, distensione addominale, eruttazioni, ipoglicemia (basso livello di glucosio nel sangue) nelle persone con diabete di tipo II (ma non limitato ad esse), flatulenza, gastroenterite e malattia da reflusso gastroesofageo (GERD). In passato è stato sospettato di causare pancreatite e può causare gastroparesi e ostruzione intestinale.[144]Tra le persone a cui è stato prescritto un agonista del recettore del GLP-1, lo 0,1% ha ricevuto una diagnosi di gastroparesi. L’1% ha ricevuto una diagnosi di gastroparesi almeno sei mesi dopo, il che equivale a un aumento del 52% del rischio di diagnosi di gastroparesi durante l’assunzione di un farmaco di questa classe.[145] Una meta-analisi del 2019 non ha indicato un rischio significativamente elevato di pancreatite acuta.[146]Secondo il sistema di segnalazione degli eventi avversi dell’FDA (FAERS), più di 150 pazienti che assumevano Ozempic hanno riportato ileo o ostruzioni intestinali dopo l’assunzione del farmaco.[147]
Confronto visivo tra stomaco sano e stomaco con gastroparesi.
L’etichetta dell’FDA statunitense per il Semaglutide contiene un boxed warning per i tumori della tiroide a cellule C nei roditori.[148] Non è noto se il Semaglutide causi tumori della tiroide a cellule C, incluso il carcinoma midollare della tiroide, nell’uomo.[149]
Tirzepatide[approvato nel 2022]
La Tirzepatide è un farmaco antidiabetico utilizzato per il trattamento del diabete di tipo II [150][151][152][153] e per la perdita di peso.[154][155] La Tirzepatide viene somministrata tramite iniezioni sottocutanee.[150][151] Viene venduta con i marchi Mounjaro per il trattamento del diabete,[150] e Zepbound per la perdita di peso.[154] La Tirzepatide è un agonista del recettore del GIP e del GLP-1.[154]
La sintesi della Tirzepatide è stata divulgata per la prima volta nei brevetti depositati da Eli Lilly and Company.[156] Questa utilizza la sintesi standard di peptidi in fase solida, con un gruppo protettivo allilossicarbonilico sulla Lisina in posizione 20 della catena lineare degli amminoacidi, consentendo una serie finale di trasformazioni chimiche in cui l’ammina della catena laterale di tale Lisina viene derivatizzata con il frammento contenente lipidi.
Per questo composto sono stati riportati processi di produzione su larga scala.[157]
La Tirzepatide è un analogo dell’ormone GIP umano con una porzione diacidica grassa C20, utilizzata per ottimizzare l’assorbimento e il metabolismo del composto.[158] La sezione diacidica grassa (acido eicosanedioico) è legata tramite un acido glutammico e due unità di acido (2-(2-aminoetossi)etossico)acetico alla catena laterale del residuo di Lisina. Questa disposizione consente un’emivita molto più lunga, prolungando il tempo tra una dose e l’altra, grazie alla sua elevata affinità con l’albumina.[159]
Quindi, la Tirzepatide è un polipeptide lineare di 39 aminoacidi che è stato modificato chimicamente mediante lipidazione per migliorarne l’assorbimento nelle cellule e la stabilità al metabolismo.[158] Ha completato la sperimentazione di fase III a livello globale nel 2021.[160][161]
La Tirzepatide ha un’affinità maggiore per i recettori GIP rispetto ai recettori GLP-1 e questo comportamento da doppio agonista ha dimostrato di produrre una maggiore riduzione dell’iperglicemia rispetto a un agonista selettivo dei recettori GLP-1.[162] Studi di segnalazione hanno riportato che la Tirzepatide imita le azioni del GIP naturale sul recettore GIP. [Studi di segnalazione hanno riportato che la Tirzepatide imita le azioni del GIP naturale sul recettore del GIP.[163] Tuttavia, sul recettore del GLP-1, la Tirzepatide mostra una predilezione per la generazione di cAMP (un messaggero associato alla regolazione del metabolismo del glicogeno, degli zuccheri e dei lipidi), piuttosto che per il reclutamento della β-arrestina. Questa combinazione di preferenza verso il recettore GIP e di proprietà di segnalazione distinte del GLP-1 suggerisce che questo agonismo distorto aumenta la secrezione di Insulina.[163] È stato riportato che la Tirzepatide aumenta i livelli di adiponectina, un’adipochina coinvolta nella regolazione del metabolismo del glucosio e dei lipidi, con un aumento massimo del 26% rispetto al basale dopo 26 settimane, al dosaggio di 10mg.[162]
GIP e GLP-1: somiglianze e differenze. La GIP è secreta dalle cellule K dell’intestino tenue prossimale (duodeno e digiuno), mentre il GLP-1 è secreto dalle cellule L dell’intestino tenue e crasso (ileo distale e colon), in seguito all’introduzione di carboidrati, trigliceridi, proteine o aminoacidi. Un’importante eccezione è rappresentata dalla glutammina, che è uno stimolatore specifico del GLP-1. GIP e GLP-1 determinano la secrezione di Insulina in modo dipendente dal glucosio. Le azioni del GLP-1 e del GIP sulla secrezione di Glucagone sono diverse: il GLP-1 sopprime il Glucagone durante l’iperglicemia, ma non in presenza di una normale concentrazione di glucosio plasmatico a digiuno, mentre il GIP può stimolare la secrezione di Glucagone a digiuno, durante l’ipoglicemia e l’iperglicemia. Per quanto riguarda il tessuto adiposo, il GLP-1 stimola la lipolisi, mentre il GIP determina un accumulo di grasso corporeo.
Negli studi preliminari finanziati dall’industria che hanno confrontato la Tirzepatide con la Semaglutide, la Tirzepatide ha mostrato un miglioramento minore delle riduzioni (2,01%-2,30% a seconda del dosaggio) nei test dell’emoglobina glicata rispetto alla Semaglutide (1,86%). [164] Una dose di 10mg si è dimostrata efficace anche nel ridurre l’insulino-resistenza, con una riduzione di circa l’8% rispetto al basale, misurata utilizzando l’HOMA2-IR (calcolato con l’Insulina a digiuno).[162] I livelli a digiuno delle proteine che legano l’IGF, come IGFBP1 e IGFBP2, sono aumentati in seguito al trattamento con Tirzepatide, aumentando la sensibilità all’Insulina.[162]
IGFBP1
Una meta-analisi del 2021 ha mostrato che, nell’arco di un anno di utilizzo clinico, la Tirzepatide è risultata superiore a Dulaglutide, Semaglutide, Degludec e Insulina glargine per quanto riguarda l’efficacia glicemica e la riduzione dell’obesità.[165]
In uno studio di fase III, in doppio cieco, randomizzato e controllato, sostenuto da Eli Lilly, adulti non diabetici con un indice di massa corporea pari o superiore a 30, o pari o superiore a 27 e almeno una complicazione correlata al peso, escluso il diabete, sono stati randomizzati a ricevere Tirzepatide sottocutanea una volta alla settimana (5mg, 10mg o 15mg) o placebo. La variazione percentuale media del peso alla settimana 72 è stata di -15,0% (intervallo di confidenza [IC] al 95%, da -15,9 a -14,2) con dosi settimanali di Tirzepatide di 5mg, -19,5% (IC al 95%, da -20,4 a -18,5) con dosi di 10mg e -20,9% (IC al 95%, da -21,8 a -19,9) con dosi di 15mg. La variazione di peso nel gruppo placebo è stata del -3,1% (95% CI, da -4,3 a -1,9).[166][167][168]
La Tirzepatide è stata approvata per uso medico nell’Unione Europea nel settembre 2022.[169][170]
La dose iniziale di Tirzepatide è 2,5mg una volta a settimana. Dopo 4 settimane, la dose deve essere aumentata a 5mg una volta a settimana. Se necessario, è possibile aumentare la dose con incrementi di 2,5mg dopo un minimo di 4 settimane con la dose in uso.
Le dosi di mantenimento raccomandate sono 5mg, 10mg e 15mg.
La dose massima è 15mg una volta a settimana.
Quando Tirzepatide viene aggiunto alla terapia esistente con Metformina e/o inibitore del co- trasportatore di sodio-glucosio 2 (SGLT2i), può essere mantenuta la dose in uso di Metformina e/o SGLT2i.
Quando Tirzepatide viene aggiunto alla terapia esistente con una sulfonilurea e/o Insulina, si può considerare una riduzione della dose di sulfonilurea o Insulina per ridurre il rischio di ipoglicemia. L’automonitoraggio della glicemia è necessario per aggiustare la dose di sulfonilurea e Insulina. Si raccomanda un approccio graduale per la riduzione dell’Insulina.
Gli studi preclinici, di fase I e clinici di fase II hanno indicato che la Tirzepatide presenta effetti avversi simili a quelli di altri agonisti del recettore GLP-1, come visto in precedenza. Questi effetti si verificano in gran parte a livello del tratto gastrointestinale.[171] I più frequentemente osservati sono nausea, diarrea e vomito, la cui incidenza è aumentata con l’entità del dosaggio (cioè la probabilità è maggiore quanto più alta è la dose). Anche il numero di pazienti che hanno interrotto l’assunzione di Tirzepatide è aumentato con l’aumentare del dosaggio: i pazienti che assumevano 15mg avevano un tasso di interruzione del 25% rispetto al 5,1% dei pazienti che assumevano 5mg e all’11,1% di quelli che assumevano Dulaglutide.[172] In misura leggermente minore, i pazienti hanno anche riferito una riduzione dell’appetito.[171] Altri effetti collaterali segnalati sono stati dispepsia, costipazione, dolore addominale, vertigini e ipoglicemia.[173][174]
Uso off-label e “ricreativo”:
Oltre ai loro usi medici, gli agonisti del GLP-1 hanno visto una massiva diffusione in ambito della perdita di peso a fini “estetici” nel Fitness e in parte nel BodyBuilding, resa popolare da influencer e celebrità.[175] I venditori del mercato nero offrono online prodotti non autorizzati che si spacciano per agonisti del GLP-1. Questa pratica è illegale sia negli Stati Uniti che in Europa, ma alcuni acquirenti si rivolgono a rivenditori non autorizzati perché non hanno la possibilità di farsi prescrivere legalmente il farmaco.[176][177][178][179][180] Gli acquirenti, ovviamente, corrono rischi dovuti a farmaci contraffatti o di qualità inferiore venduti da soggetti non autorizzati.[181]
L’uso, le modalità di applicazione e le limitazioni degli incretino-mimetici in campo “cosmetico” saranno riportate nella seconda parte…
Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87:1409–1439. [PubMed] [Google Scholar]
Nauck MA, Homberger E, Siegel EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. 1986;63:492–498. [PubMed] [Google Scholar]
Brown JC, Mutt V, Pederson RA. Further purification of a polypeptide demonstrating enterogastrone activity. J Physiol. 1970;209:57–64. [PMC free article] [PubMed] [Google Scholar]
Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab. 1995;80:952–957. [PubMed] [Google Scholar]
Vilsboll T, Agerso H, Lauritsen T, et al. The elimination rates of intact GIP as well as its primary metabolite, GIP 3–42, are similar in type 2 diabetic patients and healthy subjects. Regul Pept. 2006;137:168–172. [PubMed] [Google Scholar]
Drucker DJ. The biology of incretin hormones. Cell Metab. 2006;3:153–165. [PubMed] [Google Scholar]
Orskov C, Holst JJ, Poulsen SS, Kirkegaard P. Pancreatic and intestinal processing of proglucagon in man. Diabetologia. 1987;30:874–881. [PubMed] [Google Scholar]
Orci L, Bordi C, Unger RH, Perrelet A. Glucagon- and glicentin-producing cells. In: Lefebvre PJ, editor. Glucagon. Berlin: Springer Verlag; 1983. pp. 57–79. [Google Scholar]
Goke R, Larsen PJ, Mikkelsen JD, Sheikh SP. Distribution of GLP-1 binding sites in the rat brain: evidence that exendin-4 is a ligand of brain GLP-1 binding sites. Eur J Neurosci. 1995;7:2294–2300. [PubMed] [Google Scholar]
Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101:515–520. [PMC free article] [PubMed] [Google Scholar]
Gros R, You X, Baggio LL, et al. Cardiac function in mice lacking the glucagon- like peptide-1 receptor. Endocrinology. 2003;144:2242–2252. [PubMed] [Google Scholar]
Meier JJ, Gethmann A, Gotze O, et al. Glucagon-like peptide 1 abolishes the postprandial rise in triglyceride concentrations and lowers levels of non-esterified fatty acids in humans. Diabetologia. 2006;49:452–458. [PubMed] [Google Scholar]
Carraro-Lacroix LR, Malnic G, Girardi AC. Regulation of Na+/H+ exchanger NHE3 by glucagon-like peptide 1 receptor agonist exendin-4 in renal proximal tubule cells. Am J Physiol Renal Physiol. 2009;297:F1647–F1655. [PubMed] [Google Scholar]
Gault VA, O’Harte FP, Flatt PR. Glucose-dependent insulinotropic polypeptide (GIP): anti-diabetic and anti-obesity potential? Neuropeptides. 2003;37:253–263. [PubMed] [Google Scholar]
Tsukiyama K, Yamada Y, Yamada C, et al. Gastric inhibitory polypeptide as an endogenous factor promoting new bone formation after food ingestion. Mol Endocrinol. 2006;20:1644–1651. [PubMed] [Google Scholar]
Knop FK, Vilsboll T, Hojberg PV, et al. Reduced incretin effect in type 2 diabetes: cause or consequence of the diabetic state? Diabetes. 2007;56:1951–1959. [PubMed] [Google Scholar]
Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29:46–52. [PubMed] [Google Scholar]
Bagger JO, Knop FK, Lund A, Vestergaard H, Holst JJ, Vilsboll T. Impaired Regulation of the Incretin Effect in Patients with Type 2 Diabetes Mellitus. Diabetes. 2010;58 (Suppl 1):A369. [PubMed] [Google Scholar]
Vilsboll T, Krarup T, Deacon CF, Madsbad S, Holst JJ. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes. 2001;50:609–613. [PubMed] [Google Scholar]
Kjems LL, Holst JJ, Volund A, Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes. 2003;52:380–386. [PubMed] [Google Scholar]
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993;91:301–307. [PMC free article] [PubMed] [Google Scholar]
Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1993;36:741–744. [PubMed] [Google Scholar]
European medicines Agency. European Public Assessment Report Byetta. 2006. EMEA/H/C/698. [Google Scholar]
Amori RE, Lau J, Pittas AG. Efficacy and safety of incretin therapy in type 2 diabetes: systematic review and meta-analysis. JAMA. 2007;298:194–206. [PubMed] [Google Scholar]
Heine RJ, Van Gaal LF, Johns D, Mihm MJ, Widel MH, Brodows RG. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med. 2005;143:559–569. [PubMed] [Google Scholar]
Cvetkovic RS, Plosker GL. Exenatide: a review of its use in patients with type 2 diabetes mellitus (as an adjunct to metformin and/or a sulfonylurea) Drugs. 2007;67:935–954. [PubMed] [Google Scholar]
Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664–1669. [PubMed] [Google Scholar]
Elbrond B, Jakobsen G, Larsen S, et al. Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects. Diabetes Care. 2002;25:1398–1404. [PubMed] [Google Scholar]
Damholt B, Golor G, Wierich W, Pedersen P, Ekblom M, Zdravkovic M. An open-label, parallel group study investigating the effects of age and gender on the pharmacokinetics of the once-daily glucagon-like peptide-1 analogue liraglutide. J Clin Pharmacol. 2006;46:635–641. [PubMed] [Google Scholar]
O’Neil PM, Birkenfeld AL, McGowan B, Mosenzon O, Pedersen SD, Wharton S, et al. (August 2018). “Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial”. Lancet. 392 (10148): 637–649. doi:10.1016/S0140-6736(18)31773-2. PMID30122305. S2CID52041320.
Fox CK, Barrientos-Pérez M, Bomberg EM, Dcruz J, Gies I, Harder-Lauridsen NM, et al. (September 2024). “Liraglutide for Children 6 to <12 Years of Age with Obesity – A Randomized Trial”. The New England Journal of Medicine. doi:10.1056/NEJMoa2407379. PMID39258838.
“News details”. Novo Nordisk. Retrieved 12 September 2024.
Garber A, Henry R, Ratner R, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet. 2009;373:473–481. [PubMed] [Google Scholar]
Marre M, Shaw J, Brandle M, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with Type 2 diabetes (LEAD-1 SU) Diabet Med. 2009;26:268–278. [PMC free article] [PubMed] [Google Scholar]
Buse JB, Rosenstock J, Sesti G, et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6) Lancet. 2009;374:39–47. [PubMed] [Google Scholar]
Parks M, Rosebraugh C (March 2010). “Weighing risks and benefits of liraglutide–the FDA’s review of a new antidiabetic therapy”. The New England Journal of Medicine. 362 (9): 774–7. doi:10.1056/NEJMp1001578. PMID20164475.
Singh S, Chang HY, Richards TM, Weiner JP, Clark JM, Segal JB (April 2013). “Glucagonlike peptide 1-based therapies and risk of hospitalization for acute pancreatitis in type 2 diabetes mellitus: a population-based matched case-control study”. JAMA Internal Medicine. 173 (7): 534–539. doi:10.1001/jamainternmed.2013.2720. PMID23440284. S2CID425632.
Tibble CA, Cavaiola TS, Henry RR (May 2013). “Longer acting GLP-1 receptor agonists and the potential for improved cardiovascular outcomes: a review of current literature”. Expert Review of Endocrinology & Metabolism. 8 (3): 247–259. doi:10.1586/eem.13.20. PMID30780817. S2CID73313508.
Monami M, Dicembrini I, Nardini C, Fiordelli I, Mannucci E (February 2014). “Glucagon-like peptide-1 receptor agonists and pancreatitis: a meta-analysis of randomized clinical trials”. Diabetes Research and Clinical Practice. 103 (2): 269–275. doi:10.1016/j.diabres.2014.01.010. PMID24485345. S2CID33922845.
Christensen M, Knop FK, Holst JJ, Vilsboll T (August 2009). “Lixisenatide, a novel GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus”. IDrugs. 12 (8): 503–13. PMID19629885.
McClean PL, Hölscher C (November 2014). “Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease”. Neuropharmacology. 86: 241–58. doi:10.1016/j.neuropharm.2014.07.015. PMID25107586. S2CID24550291.
Cai HY, Yang JT, Wang ZJ, Zhang J, Yang W, Wu MN, Qi JS (January 2018). “Lixisenatide reduces amyloid plaques, neurofibrillary tangles and neuroinflammation in an APP/PS1/tau mouse model of Alzheimer’s disease”. Biochemical and Biophysical Research Communications. 495 (1): 1034–1040. doi:10.1016/j.bbrc.2017.11.114. PMID29175324.
Clinical trial number NCT00696657 for “A Randomised Controlled Clinical Trial in Type 2 Diabetes Comparing Semaglutide to Placebo and Liraglutide” at ClinicalTrials.gov
“Wegovy : Pending EC decision”. European Medicines Agency. 11 November 2021. Archived from the original on 13 November 2021. Retrieved 13 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^ Abd El Aziz M, Cahyadi O, Meier JJ, Schmidt WE, Nauck MA (April 2020). “Incretin-based glucose-lowering medications and the risk of acute pancreatitis and malignancies: a meta-analysis based on cardiovascular outcomes trials”. Diabetes, Obesity & Metabolism. 22 (4): 699–704. doi:10.1111/dom.13924. PMID31750601.
US patent 9474780, Bokvist BK, Coskun T, Cummins RC, Alsina-Fernandez J, “GIP and GLP-1 co-agonist compounds”, issued 2016-10-25, assigned to Eli Lilly and Co
Ahangarpour M, Kavianinia I, Harris PW, Brimble MA (January 2021). “Photo-induced radical thiol-ene chemistry: a versatile toolbox for peptide-based drug design”. Chemical Society Reviews. 50 (2). Royal Society of Chemistry: 898–944. doi:10.1039/d0cs00354a. PMID33404559. S2CID230783854.
“Mounjaro EPAR”. European Medicines Agency (EMA). 18 July 2022. Archived from the original on 12 December 2022. Retrieved 2 January 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Il concetto di “ricomposizione corporea” nel Fitness e nel BodyBuilding è senza dubbio considerabile come il “fattore dominante” ricercato dal momento che si tratta, molto semplicemente, del miglioramento quantitativo e qualitativo della massa contrattile (muscolo-scheletrico) a discapito della massa grassa e della ritenzione idrica extacellulare. Che si parli di “Natursl” o “Enhanced”, oltre alle variabili alimentari e allenanti vi sono quelle supplementative rappresentate, dipendentemente dalla “filosofia” scelta, da supplementi OTC e da farmaci utilizzati in ambito off-label.
Caffeina e p-Sinefrina rappresentano i lipolitici/termogenici OTC più utilizzati con un discreto margine di efficacia. Nel contesto “Enhanced”, invece, le classi di farmaci utilizzate al fine di accentuare la riduzione (direttamente o indirettamente) della massa grassa sono diverse e comprendono comunemente:
i β2-agonisti (non selettivi e selettivi) come Efedrina, Clenbuterolo e Salbutamolo;
i β3-agonisti selettivi come il Mirabegron;
gli agenti anoressizzanti con azione sui neurotrasmettitori come la Sibutramina, la Lorcaserina, l’Amfepramone e il Benfluorex;
gli anoressizzanti analoghi incretinici come la Semaglutide, il Liraglutide e il Tirzepatide;
i tiroidei Tiroxina (T4), Triiodotironina (T3) e Diiodotironina (T2);
i disaccoppianti della fosforilazione ossidativa come il 2,4-dinitrofenolo (DNP);
stimolanti il Il Peptide Natriuretico Atriale (ANP) – vedi, ad esempio, i β-bloccanti – ;
α2-antagonisti come la Yohimbina e l’α-yohimbina [Rauwolscine];
trattamenti mesoterapici a base di Fosfatidilcolina e/o Acidi Biliari.
A questo elenco, però, andrebbe aggiunta una classe di farmaci che molto poco intuitivamente ci fa pensare alla riduzione della massa grassa. Tale classe di farmaci è rappresentata dagli ACE II inibitori.
Per iniziare a comprendere del perchè questi farmaci possono rappresentare una componente funzionale nel miglioramento della composizione corporea, bisogna parlare di “Stubborn Fat” [“Grasso Testardo”]. Perchè è proprio in questa specifica e caratteristica area del tessuto adiposo che l’ACE II inibitore può contribuire alla riduzione della massa grassa.
Al fine di avere una visione di insieme più completa, è necessario trattare in modo adeguato tutte le componenti dell'”equazione”…
Tessuto adiposo e sue caratteristiche:
Il tessuto adiposo (noto anche come grasso corporeo o semplicemente grasso) è un tessuto connettivo lasso composto principalmente da adipociti.[1][2] Contiene anche la frazione vascolare stromale (SVF) di cellule tra cui preadipociti, fibroblasti, cellule endoteliali vascolari e una varietà di cellule immunitarie come i macrofagi del tessuto adiposo. Il suo ruolo non è semplicemente e solo quello di immagazzinare energia sotto forma di lipidi, ma anche di ammortizzare e isolare il corpo e rappresenta un vero e proprio organo endocrino.
Leptina
Infatti, il tessuto adiposo veniva considerato inerte dal punto di vista ormonale, ma negli ultimi anni è stato riconosciuto come un importante organo endocrino,[3] in quanto produce ormoni come Leptina, Estrogeni, Resistina e Citochine (in particolare il TNFα). Nell’obesità, il tessuto adiposo è coinvolto nel rilascio cronico di marcatori pro-infiammatori noti come adipochine, che sono responsabili dello sviluppo della sindrome metabolica, una costellazione di malattie tra cui il diabete di tipo II, le malattie cardiovascolari e l’aterosclerosi.[2][4]
Preadipociti umani sottocutanei.
Il tessuto adiposo deriva dai preadipociti e la sua formazione sembra essere controllata in parte dal gene dell’adipe. Sappiamo ormai bene che vi sono due principali tipi di tessuto adiposo, il tessuto adiposo bianco (WAT), che immagazzina energia, e il tessuto adiposo bruno (BAT), che genera calore corporeo. Il tessuto adiposo, più precisamente il tessuto adiposo bruno, è stato identificato per la prima volta dal naturalista svizzero Conrad Gessner nel 1551.[5]
Grasso Viscerale e Sottocutaneo:
Grasso Viscerale: Il grasso viscerale o addominale[6] (noto anche come grasso d’organo o grasso intra-addominale) si trova all’interno della cavità addominale, stipato tra gli organi (stomaco, fegato, intestino, reni, ecc.). Il grasso viscerale è diverso dal grasso sottocutaneo e dal grasso intramuscolare presente nei muscoli scheletrici. Il grasso nella parte inferiore del corpo, come nelle cosce e nei glutei, è sottocutaneo e non è un tessuto omogeneo, mentre il grasso nell’addome è per lo più viscerale e semi-fluido.[7] Il grasso viscerale è composto da diversi depositi adiposi, tra cui il tessuto adiposo mesenterico, il tessuto adiposo bianco epididimale (EWAT) e i depositi perirenali. Il grasso viscerale viene spesso espresso in termini di area in cm2 (VFA, visceral fat area).[8]
Da sinistra: Normale funzione dell’insulina nell’adipocita e Resistenza all’Insulina nell’adipocita.
Un eccesso di grasso viscerale è noto come obesità addominale, o “grasso della pancia”, in cui l’addome sporge eccessivamente. Nuovi sviluppi, come il Body Volume Index (BVI), sono specificamente progettati per misurare il volume addominale e il grasso addominale. L’eccesso di grasso viscerale è anche legato al diabete di tipo II,[9] all’insulino-resistenza,[10] alle malattie infiammatorie,[11] e ad altre patologie correlate all’obesità.[12] Allo stesso modo, è stato dimostrato che l’accumulo di grasso del collo (o tessuto adiposo cervicale) è associato alla mortalità.[13] Diversi studi hanno suggerito che il grasso viscerale può essere previsto da semplici misure antropometriche,[14] e predice la mortalità in modo più accurato dell’indice di massa corporea o della circonferenza vita.[15]
Gli uomini hanno maggiori probabilità di accumulare grasso nell’addome a causa delle differenze tra gli ormoni sessuali. L’estrogeno causa l’accumulo di grasso nei glutei, nelle cosce e nei fianchi delle donne.[16][17] Quando le donne raggiungono la menopausa e gli estrogeni prodotti dalle ovaie diminuiscono, il grasso migra dai glutei, dai fianchi e dalle cosce alla vita;[18] in seguito il grasso viene accumulato nell’addome.[7]
Il grasso viscerale può essere causato da un eccesso di livelli di cortisolo.[19] Almeno 10 ore MET a settimana di esercizio aerobico portano a una riduzione del grasso viscerale in chi non ha disturbi legati al metabolismo.[20] Anche l’allenamento contro-resistenza e la restrizione calorica riducono il grasso viscerale, anche se il loro effetto può non essere cumulativo.[21] Sia l’esercizio che la dieta ipocalorica causano la perdita di grasso viscerale, ma l’esercizio ha un effetto maggiore sul grasso viscerale rispetto al grasso totale. [22] L’esercizio fisico ad alta intensità è un modo per ridurre efficacemente il grasso addominale totale.[23][24] Una dieta ipocalorica combinata con l’esercizio fisico riduce il grasso corporeo totale e il rapporto tra tessuto adiposo viscerale e tessuto adiposo sottocutaneo, suggerendo una mobilitazione preferenziale del grasso viscerale rispetto al grasso sottocutaneo.[25] Il grasso addominale è fortemente soggetto alle variabili dell’Insulino-resistenza/sensibilità.
Grasso Sottocutaneo: La maggior parte del grasso non viscerale rimanente si trova appena sotto la pelle, in una regione chiamata ipoderma.[26] Questo grasso sottocutaneo non è correlato a molte delle classiche patologie legate all’obesità, come le malattie cardiache, il cancro e l’ictus, e alcune prove suggeriscono addirittura che potrebbe essere protettivo.[27] Il modello tipicamente femminile (o ginecoide) di distribuzione del grasso corporeo intorno ai fianchi, alle cosce e ai glutei è costituito da grasso sottocutaneo, e quindi rappresenta un rischio minore per la salute rispetto al grasso viscerale.[28][29]
Come tutti gli altri organi adiposi, il grasso sottocutaneo è parte attiva del sistema endocrino e secerne gli ormoni Leptina e Resistina.[26]
La relazione tra lo strato adiposo sottocutaneo e il grasso corporeo totale di una persona viene spesso modellata utilizzando equazioni di regressione. La più popolare di queste equazioni è stata creata da Durnin e Wormersley, che hanno testato in modo rigoroso molti tipi di dermoprotezione e, di conseguenza, hanno creato due formule per calcolare la densità corporea di uomini e donne. Queste equazioni presentano una correlazione inversa tra le pieghe cutanee e la densità corporea: all’aumentare della somma delle pieghe cutanee, la densità corporea diminuisce.[30]
Fattori come il sesso, l’età, le dimensioni della popolazione o altre variabili possono rendere le equazioni non valide e inutilizzabili e, a partire dal 2012, le equazioni di Durnin e Wormersley rimangono solo stime del reale livello di grassezza di una persona. Nuove formule sono ancora in fase di creazione.[30]
Gli adipociti del grasso sottocutaneo sono il target degli sforzi di manipolazione dietetica, allenante e supplementativa per ridurre al massimo la percentuale di grasso corporeo. Vi sono comunque aree di distribuzione del grasso sottocutaneo con tassi di mobilitazione lipidica differenti tra gli individui. Ed è proprio in riferimento alle aree di più difficile mobilitazione che ci si riferisce con il termina “grasso ostinato” .
Fisiologia del tessuto adiposo:
Gli acidi grassi liberi (FFA) vengono rilasciati dalla lipoproteina lipasi (LPL) ed entrano nell’adipocita, dove vengono riassemblati in trigliceridi mediante esterificazione con il glicerolo.[2] Il tessuto adiposo umano contiene circa l’87% di lipidi.[31]
Esiste un flusso costante di FFA che entrano ed escono dal tessuto adiposo.[2] La direzione netta di questo flusso è controllata dall’insulina e dalla leptina: se l’insulina è elevata, c’è un flusso netto di FFA verso l’interno e solo quando l’insulina è bassa gli FFA possono lasciare il tessuto adiposo. La secrezione di Insulina è stimolata dall’aumento della glicemia, dagli AA insulinogenici e in piccola parte dai grassi.[32]
β2-AR
Nell’uomo, la lipolisi (idrolisi dei trigliceridi in acidi grassi liberi) è controllata attraverso il settaggio equilibrato dei recettori β-adrenergici lipolitici e dell’antilipolisi mediata dai recettori α2A-adrenergici.
L’equilibrio tra β2 e α2A-AR determina le caratteristiche peculiari dell’adipocita in termini di lisi dei trigliceridi di deposito (perdita di grasso). Infatti, se l’equilibrio tende a perdersi in favore dei α2A-AR a discapito dei β2-AR ci troviamo di fronte al già prima citato “grasso testardo”.
Distribuzione degli Adrenocettori negli adipociti bianchi, bruni e beige
Gli adipociti bianchi sono il tipo di adipocita predominante nell’organismo e sono localizzati in depositi WAT distinti, caratterizzati da grasso intra-addominale (grasso viscerale che circonda gli organi interni, ovvero grasso mesenterico, perirenale e gonadico) o sottocutaneo (come il grasso inguinale). Gli adipociti bianchi immagazzinano energia (glucosio e acidi grassi) sotto forma di trigliceridi all’interno di un’unica goccia lipidica e il WAT agisce anche come organo endocrino per il rilascio di adipochine come la leptina e l’adiponectina che regolano l’omeostasi energetica dell’intero corpo (Galic, Oakhill, & Steinberg, 2010).
Differenze nella visualizzazione, nella funzione e nell’espressione dei geni firma negli adipociti bianchi, bruni e beige e l’attuale comprensione dell’espressione e della funzione degli adrenocettori (AR) nei roditori e nell’uomo. La mancata menzione di un sottotipo di adrenocettore indica che non esistono prove attuali dell’espressione/funzione della proteina recettoriale. In alcuni casi, l’evidenza funzionale si basa sull’uso di agonisti non selettivi (✦), tra cui l’Isoprenalina (Bartesaghi et al., 2015) e l’Efedrina (Carey et al., 2013) o di antagonisti (✧), tra cui la Fentolamina (Stich et al., 1999) o una combinazione di Propranololo e SR59230A per inibire tutte le risposte mediate dai β-adrenocettori (Imai et al., 2006). *L’assorbimento di 2-[18F]fluoro-2-deossiglucosio è stato misurato in risposta all’agonista selettivo dei β3-adrenocettori mirabegron nel tessuto adiposo bruno umano (Cypess et al., 2015). BA: adipocita bruno; UCP1: proteina di disaccoppiamento 1; WA: adipocita bianco
Nei roditori, tutti e tre i sottotipi di β-adrenocettori sono espressi in una serie di depositi sottocutanei e viscerali (Collins et al., 1994; Collins, Daniel, & Rohlfs, 1999; Germack, Starzec, Vassy, & Perret, 1997; Granneman, 1992; Hollenga & Zaagsma, 1989; Komai et al, 2016; Llado et al., 2002; Susulic et al., 1995), con il β3-adrenocettore che è il principale recettore responsabile della lipolisi mediata dal β-adrenocettore negli adipociti bianchi maturi. L’espressione del β-adrenocettore è influenzata anche dallo stato di differenziazione dell’adipocita bianco. L’agonista generale dei β-adrenocettori, l’Isoprenalina, ma non l’agonista altamente selettivo dei β3-adrenocettori, il CL316243, aumenta la proliferazione dei preadipociti, suggerendo un ruolo mediato dai β1-adrenocettori, mentre sia i β1-adrenocettori che i β3-adrenocettori mediano la lipolisi negli adipociti maturi (Germack et al., 1997; Klaus, Seivert, & Boeuf, 2001; Louis, Jackman, Nero, Iakovidis, & Louis, 2000; Susulic et al., 1995). È stato escluso un ruolo del β2-adrenocettore utilizzando antagonisti e agonisti selettivi del recettore.
Questi studi dimostrano collettivamente che i β-adrenocettori sono essenziali per la funzione del WAT, ma che esistono meccanismi di compensazione quando manca il β3-adrenocettore. Non ci sono prove convincenti di un contributo funzionale da parte degli α1- o α2-adrenocettori negli adipociti bianchi autentici dei roditori (Merlin, Sato, Nowell, et al., 2018). Le conoscenze sulla regolazione dell’adiponectina da parte degli adrenocettori sono meno numerose. L’adiponectina, una seconda adipochina secreta dagli adipociti bianchi e bruni, regola l’assorbimento del glucosio, la lipogenesi, la lipolisi e l’ossidazione degli acidi grassi in diversi tessuti, compreso il WAT, in modo autocrino.
Negli esseri umani, l’α1A-adrenocettore mostra una forte espressione in tutti i campioni adulti nativi, ma un’espressione trascurabile negli adipociti coltivati. Al contrario, l’mRNA per l’α1B-adrenocettore è osservato nei tessuti nativi ma anche negli adipociti differenziati di tutti i depositi, mentre l’espressione dell’α1D-adrenocettore è estremamente bassa sia nei tessuti che nelle colture primarie. L’α2A-adrenocettore mostra una forte espressione nei depositi di WAT adulto, un’espressione molto più bassa nel BAT e un’espressione bassa ma significativa nelle colture di adipociti umani maturi. L’espressione dell’α2B-adrenocettore è massima nel BAT fetale, mentre quella dell’α2C-adrenocettore è elevata nel WAT adulto e nel BAT fetale. Livelli significativi di mRNA di α2C-adrenocettori sono osservati anche negli adipociti bruni interscapolari fetali in coltura. Come accennato in precedenza, esiste un’ampia letteratura sul ruolo dei β-adrenocettori nel tessuto adiposo animale; è quindi interessante che tutti e tre i recettori siano espressi nei depositi adiposi umani nativi. Gli mRNA dei β1- e β2-adrenocettori sono presenti in tutti i depositi del BAT e del WAT, mentre l’mRNA del β3-adrenocettore è espresso principalmente nel BAT sopraclaveare adulto. Come altri marcatori termogenici, il numero di β3-adrenocettori è aumentato nel BAT sovraclaveare di un soggetto esposto al freddo (Chondronikola et al., 2016).
Rapporti precedenti hanno utilizzato la RT-PCR per dimostrare l’espressione dei β3-adrenocettori nel WAT, sebbene i segnali fossero costantemente più elevati nel BAT infantile o nel BAT perirenale (Krief et al., 1993; Lonnqvist et al., 1993; Tavernier et al., 1996). Il riscontro costante di una bassissima espressione di β3-adrenocettori nel WAT, sia da RT-PCR che da RNA-Seq, suggerisce che potrebbero esistere sottopopolazioni minori di cellule positive ai β3-adrenocettori nei depositi di WAT umano.
Le colture di adipociti derivate dalla SVF di depositi adiposi umani mostrano un’espressione trascurabile dei β3-adrenocettori, anche dopo il differenziamento in presenza di cocktail altamente adipogenici (Ding et al., 2018; Shinoda et al., 2015). L’mRNA del β1-adrenocettore è trascurabile anche negli adipociti umani primari, mentre il β2-adrenocettore è espresso nelle colture differenziate con valori medi di frammenti per kilobase per milione di reads di 1,8 (adipociti bruni sopraclavicolari) e 2,2 (adipociti bianchi sottocutanei). La mancanza di espressione dei β3-adrenocettori si verifica parallelamente a bassi livelli di mRNA per PPARGC1A, CPT1B e UCP1, tutti elementi centrali per il controllo cellulare della termogenesi. Ciò suggerisce che la differenziazione di adipociti bruni o beige termogenici è difficile da ottenere sperimentalmente negli adipociti umani primari derivati dalla SVF. Shinoda et al. (2015) hanno osservato che la differenziazione di colture clonali di adipociti bruni sopraclavicolari in presenza di 1 μM di Rosiglitazone e/o il trattamento degli adipociti maturi con 10 μM di Forskolina per 4 ore era sufficiente a indurre livelli di espressione di UCP1 simili a quelli osservati nelle biopsie scBAT native, come osservato nelle colture di adipociti bruni e beige di topo (Merlin, Sato, Chia, et al., 2018). Questo tipo di induzione potrebbe essere necessaria per promuovere l’espressione dei β3-adrenocettori, di PPARGC1A e di CPT1B.
L’espressione a basso livello dei sottotipi di β-adrenocettori è stata rilevata mediante qPCR nelle cellule staminali umane multipotenti di derivazione adiposa, con un rapporto di 3:12:1 per i β1:β2:β3-adrenocettori (Mattsson et al., 2011), ma solo gli agonisti dei β1- e β3-adrenocettori aumentano i livelli di mRNA e di proteina di UCP1 in queste cellule (Mattsson et al., 2011). Le cellule differenziate SGBS e PAZ6 sono state analizzate mediante RNA-Seq (Guennoun et al., 2015). L’espressione del β3-adrenocettore non è rilevabile nelle cellule SGBS, ma è significativa nelle cellule PAZ6 differenziate (2,5 RPKM (reads per kilobase per million mapped reads); Guennoun et al., 2015). È quindi evidente che i livelli di espressione degli adrenocettori e dei marcatori termogenici devono essere considerati in diversi sistemi modello quando si studiano potenziali agenti di “inbrunenti”.
Il WAT umano e gli adipociti bianchi dei roditori differiscono significativamente nell’espressione degli α2-adrenocettori, con un’alta espressione degli α2-adrenocettori nel WAT umano (Galitzky, Larrouy, Berlan, & Lafontan, 1990; Mauriege et al, 1991; Mauriege, Marette, et al, 1995; Mauriege, Prud’homme, Lemieux, Tremblay, & Despres, 1995), ma bassa espressione negli adipociti bianchi dei roditori (Merlin, Sato, Nowell, et al. , 2018; Valet et al., 2000). Ormai sappiamo che l’attivazione di α2-adrenocettori accoppiati a Gαi/o negli adipociti bianchi umani inibisce gli aumenti della lipolisi stimolati dalle catecolamine, contrastando così la lipolisi mediata dai β-adrenocettori (Stich et al, 1999), e gli adipociti bianchi degli esseri umani obesi presentano livelli aumentati di α2-adrenocettori, aumento di α2: β-adrenocettori e un aumento delle risposte mediate dagli α2-adrenocettori (Galitzky et al, 1990; Mauriege et al. , 1991; Mauriege, Marette, et al., 1995; Mauriege, Prud’homme, et al., 1995). Quando l’α2-adrenocettore umano è sovraespresso nel tessuto adiposo di topi KO con β3-adrenocettore, la lipolisi mediata dalla catecolamina negli adipociti bianchi è attenuata e i topi sviluppano una maggiore obesità con una dieta ad alto contenuto di grassi (Valet et al., 2000). Nonostante l’espressione significativa degli α1A- e α1B-adrenocettori nel tessuto adiposo umano nativo, non vi sono prove funzionali convincenti di un’attività diretta delle catecolamine.
“Stubborn Fat”
I due tipi di adrenocettori sopra citati, non controllano solo il metabolismo delle cellule grasse, ma anche il flusso sanguigno in entrata e in uscita da queste ultime. Di conseguenza, i β2-AR aumentano la lipolisi e il flusso sanguigno del tessuto adiposo mentre i α2A-AR inibiscono la lipolisi e il flusso sanguigno del tessuto adiposo.
Quindi, le diverse aree del grasso corporeo hanno una diversa distribuzione degli adrenorecettori β2 e α2A e questo influisce profondamente sulla capacità o meno di mobilitare e trasportare il grasso al di fuori di esse.
L’esempio più estremo è quello del grasso corporeo inferiore (fianchi e cosce), in cui è stato riscontrato un numero di recettori α2A circa 9 volte maggiore rispetto ai recettori β2. Alcune ricerche suggeriscono che il grasso addominale degli uomini ha una maggiore densità di recettori α2A (rispetto, ad esempio, al grasso viscerale), anche se non è così accentuato come per il grasso corporeo inferiore. Sebbene non sia stato studiato, è probabile che anche il grasso della parte inferiore della schiena sia relativamente resistente agli stimoli lipolitici, a causa di un numero maggiore di recettori α2A.
I dismorfismi sessuali sulla ripartizione calorica sembrano mostrare che nelle donne, dopo un pasto, può verificarsi una distribuzione calorica preferenziale nel grasso dell’area inferiore del corpo, oltre ad una ridistribuzione del grasso dalla parte superiore a quella inferiore del corpo.
Non è raro, infatti, che le donne lamentino una perdita sensibile nella parte superiore del corpo con una concomitante ed apparente peggioramento dei depositi adiposi nella parte inferiore. Una donna potrebbe mobilitare bene il grasso della parte superiore del corpo, ma immagazzinare parte di quel grasso nei depositi della parte inferiore del corpo. La parte superiore del corpo diventa più magra, quella inferiore più grassa. Questa possibilità può interessare a diverso grado anche gli uomini.
Come accennato in precedenza, oltre alle differenze nella reattività agli stimoli lipolitici, i depositi di “grasso testardo” hanno un flusso sanguigno significativamente più scarso rispetto ad altri depositi.
Alcuni studi hanno dimostrato che il flusso sanguigno nella parte inferiore del corpo può essere inferiore del 67% rispetto ad altri depositi. Il grasso viscerale ha un flusso sanguigno estremamente buono e viene mobilitato molto rapidamente.
La scarsa circolazione sanguigna ha due conseguenze importanti. In primo luogo, significa che gli ormoni trasportati dal sangue non possono raggiungere a concentrazioni ottimali le cellule adipose. In secondo luogo, un flusso sanguigno insufficiente rende più difficile far uscire il grasso mobilitato dalla cellula grassa per ossidarlo altrove.
Il motivo per cui il flusso sanguigno è così scarso non è ben definito. In parte potrebbe trattarsi semplicemente di un minor numero di vasi sanguigni, visto che gli studi di imaging ne mostrano pochi in quell’area. Inoltre, sembra che i vasi sanguigni della parte inferiore del corpo abbiano più recettori α2A che β2; ciò ha la stessa conseguenza della lipolisi. Più recettori α2A significano più vasocostrizione e meno vasodilatazione, il che si traduce in un minor flusso sanguigno.
Un fattore da tenere in considerazione è che, l’Estradiolo aumenta direttamente il numero di recettori α2A-adrenergici antilipolitici negli adipociti sottocutanei. L’aumento del numero di recettori α2A-adrenergici causa una risposta lipolitica attenuata delle Catecolamine o delle ammine simpaticomimentiche negli adipociti sottocutanei; al contrario, non è stato osservato alcun effetto degli estrogeni sull’espressione dell’mRNA dei recettori α2A-adrenergici negli adipociti del deposito di grasso intra-addominale.
Questi risultati mostrano che una cattiva gestione degli estrogeni abbassa la risposta lipolitica nel deposito di grasso sottocutaneo aumentando il numero di recettori α2A-adrenergici antilipolitici, mentre gli estrogeni non sembrano influenzare la lipolisi negli adipociti del deposito di grasso intra-addominale. Si è scoperto che questo effetto degli estrogeni è causato dal sottotipo α del recettore degli estrogeni (ERα).
Questi risultati dimostrano che una sovraespressione estrogenica attenua la risposta lipolitica attraverso la sovra-regolazione del numero di recettori α2A-adrenergici antilipolitici solo nel sottocutaneo e non nei depositi di grasso viscerale. Ciò rappresenta una spiegazione del modo in cui gli estrogeni mantengono la tipica distribuzione del grasso femminile nel sottocute, poiché gli estrogeni sembrano inibire la lipolisi solo nei depositi sottocutanei, spostando così l’assimilazione del grasso dai depositi intra-addominali a quelli sottocutanei peggiorando la situazione dei depositi di “grasso testardo” pre-esistenti e “generandone” di nuovi.
Antagonisti degli α2-AR:
Fentolamina; un α2 bloccante
Gli α2 bloccanti sono un sottoinsieme della classe dei farmaci α-bloccanti e sono antagonisti del recettore adrenergico α2. Sono utilizzati principalmente nella ricerca, avendo trovato un’applicazione clinica limitata nella medicina umana. Gli α2 bloccanti aumentano il rilascio di Noradrenalina e bloccano, per l’appunto, l’attività recettoriale degli α2-AR.
La Yohimbina, storicamente utilizzata come afrodisiaco, è talvolta impiegata in medicina veterinaria (anche se ora è stata ampiamente sostituita dall’atipamezolo) per invertire gli effetti degli α2-AR, come la Medetomidina, utilizzati come sedativi durante gli interventi chirurgici.[33]
Gli antidepressivi tetraciclici Mianserina e Mirtazapina sono α2-bloccanti , anche se la loro efficacia come antidepressivi può derivare dalla loro attività su altri siti recettoriali.
Meccanicamente, i α2-bloccanti aumentano i neurotrasmettitori adrenergici, dopaminergici e serotoninergici e inducono la secrezione di Insulina, riducendo i livelli di zucchero nel sangue.
La sospensione repentina degli α2-bloccanti può essere difficile o pericolosa, poiché la sottoregolazione globale dei neurotrasmettitori può causare sintomi di depressione e altri problemi neurologici, e l’aumento dei livelli di zucchero nel sangue insieme alla diminuzione della sensibilità all’insulina può causare in alcuni casi stati diabetici. Inoltre, può verificarsi una riduzione della microcircolazione insieme alla supersensibilità all’adrenalina in organi come il fegato.
Yohimbina e α-yohinbina
Yohimbina
Non vi è dubbio che la Yohimbina rappresenti l’α2-antagonista più usato per ridurre il grasso corporeo e, nello specifico, le zone del “grasso testardo”.
Se assunta alla dose raccomandata (≤0,2mg per kg di peso corporeo), la Yohimbina può causare nausea, dolore addominale, vertigini, nervosismo e ansia.[34]
Dosi più elevate di Yohimbina possono essere pericolose; un rapporto del 2005 ha rilevato che la Yohimbina ha il più alto tasso di effetti tossici di qualsiasi prodotto botanico.[35] Casi di ingestione di Yohimbina in eccesso hanno suggerito che l’ansia, l’ipertensione (pressione alta), la tachicardia (frequenza cardiaca elevata), le aritmie e l’agitazione sono tra gli effetti collaterali più gravi di questo composto.[35]
La Yohimbina è un α2-antagonista adrenergico selettivo. In altre parole, ha come bersaglio e inattiva una classe di recettori del sistema nervoso che risponde al neurotrasmettitore Noradrenalina.[36] L’antagonismo dei recettori α2 aumenta il rilascio di Noradrenalina da parte del sistema nervoso simpatico, causando gli effetti stimolanti e “iperadrenergici” della Yohimbina.
La Yohimbina inibisce anche l’attività dei recettori α2 sulle cellule adipose, dove la Noradrenalina agisce normalmente per sopprimere il rilascio di grasso. L’inibizione dell’effetto antilipolitico della Noradrenalina consente una maggiore lipolisi (e conseguente ossidazione lipidica).[37]
Dosi giornaliere totali di 0,2mg/kg di peso corporeo sono state utilizzate con successo per aumentare la mobilitazione lipidica dai depositi di “grasso testardo” e la successiva ossidazione dei grassi senza implicazioni significative sui parametri cardiovascolari come la frequenza cardiaca e la pressione sanguigna. Ciò si traduce in un dosaggio giornaliero totale di:
14 mg per una persona di 68 kg
18 mg per una persona di 91 kg
22 mg per una persona di 113 kg.
Queste dosi totali giornaliere si riferiscono all’uso di Yohimbina come unico agente con azione riduttiva sulla attività dei recettori α2. Tali dosaggi vengono spesso suddivise e assunte in due o quattro dosi nel corso della giornata. Ad esempio, una persona di 68 kg potrebbe assumere 7mg due volte al giorno (lontano dai pasti) per raggiungere una dose totale di 14mg.
Nota: non tutti i soggetti sono in grado di tollerare la “dose piena” ricavata dalla sopra citata formula. In quel caso, l’utilizzatore mantiene la tose tollerabile raggiunta.
Rauwlscina
Se si considera lo stesso recettore α2, la Yohimbina sembra avere una selettività per la subunità α2C piuttosto che per la A o la B; la selettività è compresa tra 4 e 15 volte,[38] mentre la Rauwolscina [α-yohimbina] sembra non essere selettiva tra queste tre subunità.[39][38] La Rauwlscina sembra essere efficace a livello del recettore quanto la Yohimbina ma con una emivita di circa 5h contro i 30 minuti della prima emivita della Yohimbina.[40]
Il fatto che la Yohimbina è selettiva per la subunità α2C più che per altre subunità, compresa l’importante A, se parliamo di α2-AR adipocitari, la sua efficacia risulta moderatamente ridotta per la riduzione del “grasso testardo”, sebbene la subunità α2C sia ad un certo grado espressa anche nel WAT; o per lo meno lo è se utilizzata come unico agente interferente l’attività adipocitaria dei α2-AR.
Introduzione agli ACE II inibitori:
Captopril
Leonard T. Skeggs e i suoi colleghi (tra cui Norman Shumway) scoprirono l’ACE [Inibitori dell’enzima di conversione dell’angiotensina] nel plasma nel 1956.[41] Le scoperte avvenute nel corso di un annosa ricerca hanno portato allo sviluppo del Captopril, il primo ACE-inibitore attivo per via orale, nel 1975.[42]
Bradichinina
Gli ACE inibitori inibiscono l’attività dell’enzima di conversione dell’angiotensina, un componente importante del sistema renina-angiotensina che converte l’angiotensina I in angiotensina II e idrolizza la bradichinina.[43] Pertanto, gli ACE inibitori diminuiscono la formazione di angiotensina II, un vasocostrittore, e aumentano il livello di bradichinina, un vasodilatatore peptidico.[43] Questa combinazione è sinergica nell’abbassare la pressione sanguigna.
Gli ACE-inibitori riducono l’attività del sistema Renina-Angiotensina-Aldosterone (RAAS) come evento eziologico (causale) primario nello sviluppo dell’ipertensione nelle persone con diabete mellito, come parte della sindrome da insulino-resistenza o come manifestazione di una malattia renale.[44][45]
Il sistema renina-angiotensina-aldosterone è un importante meccanismo di regolazione della pressione sanguigna. I marcatori di squilibrio elettrolitico e idrico nell’organismo, come l’ipotensione, la bassa concentrazione di sodio nel tubulo distale, la diminuzione del volume sanguigno e l’elevato tono simpatico, innescano il rilascio dell’enzima renina dalle cellule dell’apparato juxtaglomerulare del rene.
Renina
La renina attiva un proormone circolante derivato dal fegato, l’angiotensinogeno, mediante scissione proteolitica di tutti i suoi residui aminoacidici, tranne i primi dieci, noti come angiotensina I. L’ACE (enzima di conversione dell’angiotensina) rimuove quindi altri due residui, convertendo l’angiotensina I in angiotensina II. L’ACE si trova nella circolazione polmonare e nell’endotelio di molti vasi sanguigni.[46] Il sistema aumenta la pressione sanguigna aumentando la quantità di sale e acqua trattenuta dal corpo, sebbene l’angiotensina II sia anche un potente vasocostrittore.[47]
Struttura dell’Angiotensina I e II
Gli ACE-inibitori sono stati inizialmente approvati per il trattamento dell’ipertensione e possono essere utilizzati da soli o in combinazione con altri farmaci antipertensivi. In seguito, si sono rivelati utili per altre malattie cardiovascolari e renali[48], tra cui:
Complicanze renali del diabete mellito (nefropatia diabetica), grazie alla riduzione della pressione arteriosa e alla prevenzione del danno da iperfiltrazione glomerulare[51].
Angiotesina II e tessuto adiposo:
Noradrenalina
L’angiotensina II determina, tra le atre cose, un aumento del rilascio di catecolamine (Noradrenalina), della sensibilità alle catecolamine e della loro attività.[52]
L’angiotensina II può essere prodotta dal tessuto adiposo umano; a questo proposito, l’angiotensinogeno e gli enzimi coinvolti nella sua conversione in Ang II, nonché le vie RAS (renina, enzima di conversione dell’angiotensina: ACE) e non RAS (catepsina D, catepsina G) sono espressi nel tessuto adiposo umano. Inoltre, anche i recettori dell’Ang II sono espressi nel tessuto adiposo, il che suggerisce un ruolo locale di questo ormone nella regolazione dell’adipogenesi, del metabolismo lipidico e nella patogenesi dell’obesità28,48. L’influenza dell’Ang II sugli adipociti è mediata dall’attivazione dei recettori АТ1 e АТ2, coinvolgendo diversi sistemi di trasduzione del segnale, tra cui le risposte Са 2+, la proliferazione e la differenziazione cellulare, l’accumulo di trigliceridi, l’espressione dei geni delle adipochine e la secrezione di queste ultime [53]. L’angiotensina II ha anche un effetto anti-adipogenico, riducendo la differenziazione delle cellule pre-adipose umane [54]. Pertanto, questo ormone potrebbe rappresentare un fattore protettivo contro l’espansione incontrollata del tessuto adiposo [55].Questo effetto anti-adipogenico dell’Ang II è stato osservato anche nel grasso omentale di esseri umani affetti da obesità, con la partecipazione della via della chinasi regolata dal segnale extracellulare/1,2 (ERK/1,2) e la fosforilazione del recettore gamma attivato dal proliferatore del perossisoma (pPARG) [56]. Durante questo processo, l’origine dell’Ang II può essere sia da RAS che da vie non RAS; queste ultime potrebbero essere più importanti in questo processo [57]. Tuttavia, oltre a questo effetto, l’Ang II può aumentare il contenuto di trigliceridi e l’attività di due enzimi lipogenici (FAS: sintasi degli acidi grassi e GPDH: glicerolo-3-fosfato deidrogenasi) in colture primarie di cellule adipose umane, suggerendo un controllo dell’adiposità attraverso la regolazione della sintesi e dell’immagazzinamento dei lipidi negli adipociti [58]. L’Ang II regola anche il flusso sanguigno regionale verso il tessuto adiposo e le dimensioni e il numero delle cellule grasse [59]. Queste scoperte sono state confermate dal blocco sperimentale dell’Ang II, che influenza direttamente il peso corporeo e l’adiposità [60].
Effetti adipogenici e anti-adipogenici del sistema renina-angiotensina (RAS). La produzione locale di angiotensina II (Ang II) nel tessuto adiposo è coinvolta nella regolazione dell’adipogenesi e del metabolismo lipidico. L’Ang II ha un effetto anti-adipogenico riducendo la differenziazione adipogenica delle cellule pre-adipose umane con la partecipazione di ERK e pPARG. L’Ang II può anche aumentare il contenuto di trigliceridi negli adipociti attivando due enzimi lipogenici, FAS e GPDH. Questo effetto anti-adipogenico dell’Ang II può essere regolato. L’Ang II può essere catabolizzato dall’ACE2 adiposo per formare l’Ang 1-7 che interagisce con i recettori dell’Ang 1-7 (Mas) sugli adipociti, attivando la PI3K/Akt e l’inibizione delle vie MAPK chinasi/ ERK e inducendo un effetto inibitorio nell’Ang II/AT1 anti-adipogenico, promuovendo l’adipogenesi. AT1: Recettore-1 dell’angiotensina II; AT2: Recettore-2 dell’angiotensina II; RAS: Renin Angiotensin System; Cathep D, G: Cathepsin D, Cathepsin G; ACE1: angiotensin-converting enzyme-1; ACE2: angiotensin-converting enzyme-2; Ang 1-7: Angiotensina 1-7; ERK: extracellular signal-regulated kinase; pPARG: phosphorylated peroxisome proliferator-activated receptor gamma; FAS: fatty acid synthase; GPDH: glicerolo-3-fosfato deidrogenasi; MAPK chinasi/ERK: mitogen-activated protein kinases/extracellular signal-regulated kinases; PI3K/Akt: fosfatidilinositolo 3-chinasi/proteina chinasi B.
È stata documentata anche la regolazione autocrina dell’Ang II durante l’adipogenesi. L’angiotensina II può essere catabolizzata nei tessuti adiposi dall’enzima adiposo di conversione dell’angiotensina 2 (ACE2) per formare l’Ang 1-7. La regolazione autocrina del sistema angiotensinico locale implica la coespressione dei recettori dell’Ang II (AT1 e AT2) e dei recettori dell’Ang 1-7 (Mas) sugli adipociti. L’attivazione del recettore Mas da parte dell’Ang 1-7 ha un effetto contrario all’effetto anti-adipogenico dell’Ang II, inducendo l’adipogenesi attraverso l’attivazione delle vie PI3K/Akt e l’inibizione delle vie MAPK chinasi/ERK [61] . In questo contesto, la regolazione autocrina dell’asse Ang II/AT1-ACE2-Ang 1-7/Mas durante l’adipogenesi è in grado di produrre ormoni e citochine che promuovono l’infiammazione, l’accumulo di lipidi, l’IR e le componenti del RAS, che si attivano in presenza di obesità come meccanismi chiave correlati all’obesità dell’ipertensione e di altre componenti della sindrome cardiometabolica [62].
Angiotesina II e α2A-AR
Una caratteristica di particolare interesse in riferimento all’Angiotesina II è il fatto che sia un polipeptide necessario per l’espressione di alcuni recettori α2 (ma non di tutti). Ciò significa che senza l’Angiotensina II i recettori α2 non possono essere sviluppati in alcune cellule. Di conseguenza, se sottoregoliamo l’Angiotensina II, prodotta naturalmente dall’organismo, il normale rinnovamento dei recettori α2 non avverrà. Bisogna capire che in ogni cellula c’è un costante rinnovamento recettoriale. Bloccando la formazione di un tipo specifico di recettore in una cellula (ad esempio i recettori α2), dopo un po’ di tempo non ci saranno più recettori α2 in questa cellula. I vecchi recettori saranno completamente degradati e avremo impedito alla nuova generazione di recettori di sostituire quelli vecchi.
Attività dell’Angiotesina II a livello dei α2A-AR e del Recettore dell’Angiotesina II dell’adipocita del WAT
Quindi, sotto-regolazione marcata dei α2 recettori . Il problema principale è se questa azione dell’Angiotensina II avviene nelle cellule adipose. L’Angiotensina II agisce solo sui recettori α2 che rispondono a due condizioni:
Sembra avere il massimo effetto sui recettori α2 del sottotipo “A”. Ciò è positivo, poiché sono proprio questi recettori a trovarsi nelle cellule adipose. Quindi, la prima condizione è soddisfatta.
L’Angiotensione II agisce solo sulle cellule ricche di recettori α2 e di recettori dell’Angiotensina II. Sappiamo già che le cellule adipose sono molto ricche di recettori α2. Da tempo i ricercatori sanno anche che le cellule adipose sono ricche di recettori dell’Angiotensina II.
Il punto chiave da ricordare è che nelle cellule grasse l’Angiotensina II è necessaria perché i recettori α2 si rinnovino normalmente. Se impediamo in qualche modo la formazione di Angiotensina II, causeremo grossi problemi nel rinnovo dei recettori α2A nelle cellule adipose.
Quindi, tutto ciò che occorre fare è alterare la produzione di Angiotensina II attraverso l’uso principale di ACE II inibitori. Nel giro di poche settimane il numero di recettori α2 diminuirà sensibilmente.
Quinapril
L’uso di ACE II inibitori ha quindi il potenziale di attenuare la sensibilità agli α2-adrenocettori negli adipociti umani. L’effetto del Quinapril, un ACE II inibitore lipofilo, è stato maggiore di quello dell’Enalapril [www.ncbi.nlm.nih.gov/pmc/articles], un ACE II inibitore idrofilo. Gli ACE II inibitori lipofili possono avere un effetto vasodilatatore più potente rispetto agli ACE II inibitori idrofili. La concentrazione di Angiotensina II nei tessuti piuttosto che nel plasma può contribuire alla sensibilità e il numero degli α2-adrenocettori.
Enalapril
È stato riportato che l’ACE inibitore lipofilo, Quinapril, riduce la concentrazione tissutale di Angiotensina II in misura maggiore rispetto all’ACE inibitore idrofilo, Enalapril, da 5 a 24 ore dopo una singola somministrazione orale nei ratti. I tempi di raggiungimento della concentrazione plasmatica massima del Quinapril e del suo metabolita attivo sono stati di 2-3 ore [63, 64]. Pertanto, per esaminare più chiaramente la cosa, ciascun farmaco è stato somministrato 22 e 3 ore prima dell’esame. Entrambi gli ACE inibitori hanno soppresso le attività plasmatiche dell’ACE per oltre il 90%. Questo risultato conferma i precedenti risultati ottenuti in soggetti giapponesi [65]. Sebbene la soppressione dell’attività dell’enzima convertitore dell’Angiotensina nel plasma e la pressione arteriosa sistemica non differissero tra i due farmaci, l’attenuazione della sensibilità degli α-adrenocettori alla Fenilefrina era maggiore nei soggetti trattati con Quinapril rispetto a quelli trattati con Enalapril. Le osservazioni e i rapporti precedenti [66] suggeriscono che la concentrazione di Angiotensina II nei tessuti piuttosto che quella nel plasma può contribuire alla sensibilità dei recettori α-adrenergici nei vasi ed in altri tessuti come quello adiposo. Inoltre, l’ACE inibitore lipofilo può essere più potente dell’ACE inibitore idrofilo. Infatti, il Quinapril ha attenuato la risposta vasopressore della Fenilefrina più dell’Enalapril e l’intervallo di confidenza del 95% per le differenze di ED50 tra Enalapril e Qinapril è stato di 31,1-397,5. Sebbene l’entità dell’attenuazione della sensibilità dei recettori α-adrenergici indotta dalla soppressione dell’ACE tissutale con Quinapril fosse varia, ciò è coerente con un altro esperimento in vitro [67].
Quando osservata, la concentrazione di Noradrenalina nel siero durante il riposo a letto non è cambiata prima e dopo la somministrazione del farmaco ACE inibitore. Rapporti precedenti hanno dimostrato che gli ACE inibitori attenuano il deflusso del nervo simpatico negli animali e nell’uomo [68, 69]. Negli studi in cui non è stato applicato alcun carico al sistema nervoso simpatico, non è stato possibile rilevare alcun cambiamento nel flusso simpatico indotto dagli ACE inibitori.
Applicazione degli ACE II inibitori nel trattamento del “grasso testardo”:
La genesi dell’uso degli ACEI come PEDs
Daniel (“Dan”) Duchaine
Nonostante il potenziale maggiore nella sotto-regolazione degli α2A-AR attribuita agli ACE inibitori con caratteristiche prettamente lipolifiche, la molecola appartenente a questa classe di farmaci maggiormente utilizzata per tale scopo e da più tempo è il Captopril. Questo storico ACE II inibitore mostra però caratteristiche idrofile. Certo, la sua maggiore diffusione è legata senza dubbio agli anni dalla sintesi e immissione nel circuito farmaceutico della molecola, ma anche, e soprattutto, al suo lancio come PEDs da parte, tra i primi, di Dan Duchaine (1952-2000).
Le proprietà potenziali sulla composizione corporea del Captopril vennero individuare per la prima volta in alcune atlete interessate ad assumere un farmaco che le desse un miglioramento della composizione corporea ma senza virilizzazione. Così quella divenne l’occasione giusta per testare il Captopril. La dieta delle atlete non venne cambiata. Le atlete hanno continuato per un paio di mesi ad assumere il Captopril come unico farmaco. Avevano migliorato leggermente il trofismo, ma non molto. Ciò che però colpì i “pionieri della preparazione” fu il fatto che avevano perso grasso in aree in cui prima non erano riuscite a perderlo in modo significativo.
Approfondendo le caratteristiche della molecola attraverso la consultazione di testi accademici reperiti alla biblioteca medica, scoprirono che la relazione tra il Captropril e i recettori α2.
Con il procedere del tempo e le sperimentazione dose-tempo nell’applicazione del Captopril (ma non solo), si è notato che il farmaco poteva rendere possibile la riduzione totale della dose di Yohimbina migliorando notevolmente la compliance dell’utilizzatore.
Sappiamo, infatti, che la Yohimbina presenta una selettività maggiore per i recettori α2C piuttosto che ai sottogruppi “A” e “B”. Questa caratteristica risulta limitativa nell’azione ricercata nella Yohimbina come α2-antagonista adipocitario. L’inserimento del Captopril [o di altro ACE II inibitore] permette di 1) ridurre sensibilmente il numero di α2A-AR nell’adipocita e 2) di permettere, a dosaggio di 1/2 fino a 1/3, un legame antagonista da parte della Yohimbina nei confronti degli α2-AR rimasti. L’uso della α-yohimbina, non presentando tale affinità selettiva, migliora sensibilmente questo effetto sinergico.
Situazione adipocitaria in fisiologia con attività catecolaminergica a livello degli adrenocettori nel adipocita; Impatto sulla attività adrenorecettoriale con somministrazione di Yohimbina; Impatto sulla densità/numero adrenorecettoriale con somministrazione di un ACE II inibitore [Captopril]; Impatto additivo sulla densità, numero, funzionalità e attivazione adrenorecettoriale con somministrazione di Yohimbina e un ACE II inibitore [Captopril].
Le limitazioni degli ACE II inibitori
Il Captopril [e in generale gli ACE II inibitori] non è un farmaco che manifesta rapidamente i suoi effetti dal punto di vista estetico. Bisogna ricordare che la regolazione degli α2-AR richiede almeno due mesi prima di diventare significativa.
È necessario seguire una dieta ipocalorica per vedere ottimi risultati in termini di perdita di grasso ostinato. Abbiamo detto, infatti, che i recettori α2-AR impediscono la normale perdita di grasso la dose si presentano in maggiori concentrazioni. Questo non significa, però, che si perderà automaticamente grasso di deposito solo perché si è ridotto il numero di recettori α2. Significa solo che la perdita di grasso ostinato indotta dalla dieta ipocalorica sarà più “facile”. Avrà un effetto permissivo sulla perdita di grasso ostinato, consentendo di ridurre i depositi adiposi con un rapporto di α2-AR più elevato.
L’ultima limitazione è che esiste ancora una linea di difesa per le cellule adipose e la conservazione delle riserve lipiche. Eliminando parzialmente la linea di difesa rappresentata dagli α2-AR, se ne attiva una nuova costituita da recettori antilipolitici chiamati peptide YY, anch’essi localizzati sulle cellule adipose. Ciò significa che la riduzione del livello dei recettori α2-AR permetterà di perdere più grasso ostinato di quanto sarebbe stato normalmente possibile, ma le limitazioni genetiche saranno sempre presenti.
Ma l’uso di Captopril [o altro ACE II inibitore] può permettere di fare un grande passo avanti nella giusta direzione se l’obbiettivo è una marcata riduzione della body fat, soprattutto le aree ostinate.
Esempi applicativi degli ACE II inibitori per il trattamento del “Stubborn Fat”
Nell’approccio protocollare di base, e se prendiamo come esempio di ACE II inibitore il Captopril:
Captopril = 50mg/die [da raggiungere con gradualità e aumenti giornalieri di 6,25mg];
Yohimbina = 5-10mg/die [dose da raggiungere con aumenti giornalieri (pari a 2,5mg) e test della sensibilità ];
α-yohimbina = 3/5mg die [dose da raggiungere con aumenti giornalieri (pari a 0,5mg) e test della sensibilità ].
Nell’approccio protocollare intermedio:
Captorpil = 50-75mg/die [da raggiungere con gradualità e aumenti giornalieri di 6,25mg];
Yohimbina = 10mg/die [dose da raggiungere con aumenti giornalieri (pari a 2,5mg) e test della sensibilità ];
α-yohimbina = 5-6mg die [dose da raggiungere con aumenti giornalieri (pari a 0,5mg) e test della sensibilità ];
T3 = 25mcg/die [dose da raggiungere con aumenti giornalieri (pari a 12,5mcg) e controllo ematico del FT3].
Nell’approccio protocollare avanzato:
Captorpil = 100mg/die [da raggiungere con gradualità e aumenti giornalieri di 6,25mg];
Yohimbina = 0.2mg/Kg/die [dose da raggiungere con aumenti giornalieri (pari a 2,5mg) e test della sensibilità ];
α-yohimbina = 0.1mg/Kg/die [dose da raggiungere con aumenti giornalieri (pari a 0,5mg) e test della sensibilità ];
T3 = 50mcg/die [dose da raggiungere con aumenti giornalieri (pari a 12,5mcg) e controllo ematico del FT3];
Salbutamolo = 8-12mg/die [dose da raggiungere con aumenti ogni 1-2 giorni (pari a 2mg)];
Alternativa: Clenbuterolo = 1mcg/Kg/die (range 40-80mcg) [dose da raggiungere con aumenti ogni 2 giorni (pari a 10-20mcg) e test della sensibilità/tolleranza];
Nedbivololo = 5mg/die [dose di partenza 2,5mg/die e valutazione della tolleranza].
*Nota bene: Nessuno dei protocolli sopra esposti rappresenta un indicazione d’uso o una prescrizione medica di applicazione. Tali informazioni SONO AD ESCLUSIVO SCOPO ESEMPLIFICATIVO!
Effetti collaterali degli ACE II inibitori
pressione bassa;
tosse. Un altro possibile effetto avverso specifico degli ACE-inibitori, ma non di altri bloccanti del RAAS, è l’aumento del livello di bradichinina. La tosse secca persistente è un effetto avverso relativamente comune che si ritiene sia associato all’aumento dei livelli di bradichinina prodotto dagli ACE inibitori, anche se il ruolo della bradichinina nella produzione di questi sintomi è stato contestato. Tuttavia, molti casi di tosse in persone che assumono ACE inibitori potrebbero non essere dovuti al farmaco stesso. Alcuni (0,7%) sviluppano angioedema a causa dell’aumento dei livelli di bradichinina. Può esistere una predisposizione genetica. ;
iperkaliemia. Il potassio elevato nel sangue è un’altra possibile complicazione del trattamento con un ACE-inibitore, dovuta al suo effetto sull’aldosterone. La soppressione dell’angiotensina II porta a una diminuzione dei livelli di aldosterone. Poiché l’aldosterone è responsabile dell’aumento dell’escrezione di potassio, gli ACE-inibitori possono causare una ritenzione di potassio. Alcune persone, tuttavia, possono continuare a perdere potassio durante l’assunzione di un ACE-inibitore. È necessario un attento monitoraggio dei livelli di potassio nei soggetti in trattamento con ACE-inibitori che sono a rischio di iperkaliemia.;
cefalea;
vertigini;
affaticamento;
nausea e compromissione renale. I soggetti che iniziano la terapia con un ACE-inibitore presentano di solito una modesta riduzione della velocità di filtrazione glomerulare (eGFR). Tuttavia, la riduzione può essere significativa in condizioni di preesistente ridotta perfusione renale, come stenosi dell’arteria renale, insufficienza cardiaca, malattia renale policistica o deplezione di volume. Una moderata riduzione della funzione renale, non superiore al 30% di aumento della creatinina sierica, che si stabilizza dopo una settimana di trattamento. La riduzione del eGFR è un problema soprattutto se il paziente assume contemporaneamente un FANS e un diuretico. Quando i tre farmaci vengono assunti insieme, il rischio di sviluppare un’insufficienza renale aumenta notevolmente.
Una rara reazione allergica grave può colpire la parete intestinale e causare secondariamente dolore addominale.
Ma gli ARB/Sartani possono essere un sostituto agli ACE II inibitori per lo scopo qui discusso?
Telmisartan
Sono circa vent’anni che si è scoperto che il Telmisartan, un Bloccante del Recettore dell’Angiotensina II (ARB) approvato per il trattamento dell’ipertensione, è anche un agonista parziale di PPARγ.[70-71] Mentre gli agonisti completi di PPARγ, come il Rosiglitazone e il Pioglitazone, promuovono l’aumento di peso alterando la distribuzione del grasso e la differenziazione degli adipociti, gli agonisti parziali (agonisti/antagonisti misti) di PPARγ possono avere la capacità di ritardare l’aumento di peso promuovendo al contempo la differenziazione degli adipociti.[72] Ad esempio, è stato scoperto che il Telmisartan può promuovere la differenziazione degli adipociti ma anche attenuare l’aumento di peso, migliorando al contempo il metabolismo del glucosio e dei lipidi nei ratti alimentati con una dieta ad alto contenuto di grassi e carboidrati.[70] Sharma et al[73] hanno riportato che il blocco del recettore dell’angiotensina II di tipo 1, di per sé, può promuovere la differenziazione degli adipociti e hanno proposto che questo possa contribuire agli effetti antidiabetici degli antagonisti del recettore dell’angiotensina II. Non è noto se molecole bifunzionali come il Telmisartan, che attivano PPARγ e bloccano il recettore dell’angiotensina II, esercitino effetti diversi sulle dimensioni degli adipociti e sui determinanti primari del peso corporeo rispetto ai normali bloccanti del recettore dell’angiotensina, come il Valsartan, che non hanno la capacità di attivare PPARγ.
Valsartan
Negli studi si è scoperto che il Telmisartan, ma non il Valsartan, aumenta l’espressione dei geni di un fattore di trascrizione nucleare (TFAM) che regola la funzione mitocondriale e di una proteina mitocondriale (MTCO1) coinvolta nella fosforilazione ossidativa. Rispetto agli agonisti totali convenzionali di PPARγ, come i Tiazolidinedioni, gli agonisti parziali del PPARγ, come il Telmisartan, possono avere la capacità di reclutare in modo preferenziale alcuni coattivatori trascrizionali che sono particolarmente importanti nella regolazione dei geni che controllano la funzione mitocondriale e il metabolismo energetico.[74-75] Ad esempio, gli agonisti parziali sembrano reclutare preferenzialmente il coattivatore 1-α di PPARγ, un coattivatore trascrizionale noto per stimolare l’espressione di TFAM, che, a sua volta, può aumentare l’espressione dei geni mitocondriali (ad esempio, MTCO1) e, in ultima analisi, la biogenesi mitocondriale.[75-76] Sebbene i precisi meccanismi cellulari e molecolari che mediano i robusti effetti del Telmisartan sul peso corporeo, sul dispendio energetico e sul metabolismo dei grassi rimangano da chiarire, gli studi sul reclutamento del coattivatore PPARγ e sull’espressione dei geni target, nonché sul numero, la struttura e la funzione dei mitocondri, potrebbero rappresentare aree di indagine potenzialmente fruttuose in futuro.
Ciò che si è anche notato con gli ARB, ma soprattutto con il Telmisartan, è che ha una azione sulla distribuzione del grasso più che sulla sua riduzione sistemica. Infatti, il Telmisartan ha mostrato di indurre la riduzione del grasso viscerale ma senza cambiamenti statistici sui deposito sottocutanei. Le più recenti review che hanno esaminato l’effetto del Telmisartan sulla condizione metabolica e composizione corporea dei pazienti trattati, hanno evidenziato che i risultati suggeriscono che questo sartano influisce sulla distribuzione del grasso, inducendo una riduzione del grasso viscerale, e quindi potrebbe essere utile nei pazienti ipertesi con obesità/sovrappeso, sindrome metabolica o intolleranza al glucosio.
Anche i dati aneddotici di un certo valore e design suggeriscono uno “spostamento” nell’equilibrio di mobilitazione delle riserve di grasso verso la perdita dei depositi viscerali invece di quelli sottocutanei. Ed è per tale motivo che diversi preparatori ne evitino l’uso sotto gara.
Questo “effetto shift” sul bilancio della mobilitazione delle riserve di grasso dal grasso sottocutaneo ad una prevalenza del viscerale si manifesta in modo significativo nel range di dosaggio di 80-160mg/die.
PPARγ
L’attività come agonista parziale del PPARγ è il motivo principale per il quale in Telmisartan agisce sul metabolismo lipidico adipocitario. Si è affermato che coloro i quali vogliono bypassare il problema dello shift della mobilitazione adiposa possono farlo assumendo l’Oleuropeina. Ora, non vi è nulla di certo e poco che superi la sottile linea tra ipotesi e dato realmente misurato, ma alcuni, soprattutto coloro i quali mal tollerano gli aumenti di bradichinina dati dagli ACE II inibitori, inseriscono questo supplemento erboristico nel tentativo di risolvere la sopra citata limitazione.
Peccato, però, che grazie a questa attività di agonista parziale del PPARγ, il Telmisartan può ridurre lo stoccaggio dei trigliceridi negli adipociti durante una dieta ipercalorica. In topi trattati per 28 giorni con ARB e ACE I, si è osservato un inferiore accumulo adiposo, minor peso corporeo, miglior controllo sull’assunzione di cibo rispetto ai topi non trattati con una dieta ad alto contenuto lipidico.
Nonostante, in teoria, l’effetto sul “grasso testardo” possa essere trattato anche attraverso il blocca del recettore dell’Angiotesina II, i dati a nostra disposizione ci mostrano una superiorità di azione e versatilità legata agli ACE II inibitori. L’uso di ARB, in particolar modo del Telmisartan, potrebbe avere un applicazione logica (se non si parla di soggetti obesi o in sovrappeso) nel gestione del grasso corporeo durante le fasi di ipercalorica, ad un dosaggio ipotetico di 40-80mg/die, al fine di ridurre l’accumulo adiposo e migliorare la qualità complessiva del peso raggiunto in Bulk.
Effetti collaterali degli ARB:
tachicardia e bradicardia (battito cardiaco accelerato o lento);
ipotensione (pressione sanguigna bassa);
edema (gonfiore di braccia, gambe, labbra, lingua o gola, quest’ultimo con conseguenti problemi di respirazione);
potenziale manifestazione di reazioni allergiche;
infezioni del tratto respiratorio superiore;
diarrea;
mal di schiena;
problemi renali;
iperkalemia.
Conclusioni:
Abbiamo visto come gli adrenocettori svolgono un ruolo importante nella biologia e nella fisiologia del tessuto adiposo, che comprende la regolazione della sintesi e dell’immagazzinamento dei trigliceridi (lipogenesi), la degradazione dei trigliceridi immagazzinati (lipolisi), la termogenesi (produzione di calore), il metabolismo del glucosio e la secrezione di ormoni derivati dagli adipociti che possono controllare l’omeostasi energetica dell’intero corpo. Questi processi sono regolati dal sistema nervoso simpatico attraverso l’azione di diversi sottotipi di adrenocettori espressi nei depositi di tessuto adiposo. In questa disamina, abbiamo evidenziato il ruolo dei sottotipi di adrenocettori negli adipociti bianchi, bruni e beige, e nel tessuto adiposo “testardo” ed abbiamo approfondito il ruolo potenziale degli ACE II inibitori nella modulazione sottoregolativa dell’attività degli α2-AR e l’impatto che questo può avere sul miglioramento della composizione corporea. Sono stati anche descritti gli effetti riscontrabili, nel medesimo contesto e fine, dei Sartani con le differenze tra l’applicabilità di questi confronto a quella degli ACE II inibitori.
Mentre il potenziale degli ACE II inibitori di migliorare la perdita di massa grassa in specie a carico dei depositi con una ratio sfavorevole tra α2:β2-AR, permettendo un importante sgravio sui dosaggi di α2-antagonisti, risulta un dato importante per la pianificazioni della preparazione alla gara, il potenziale effetto di riduzione del accumulo lipidico per attività di agonista parziale del PPARγ dato dal Telmisartan amplifica le applicazioni potenziali dei Sartani per il miglioramento della qualità del peso guadagnato in fase Bulk.
Ricordo, in fine, che tutto ciò che è stato detto è informazioni prettamente scientifica e non rappresenta in nessun modo un incitamento all’uso di farmaci fuori dalle linee di prescrizioni.
Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G (May 2001). “Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance”. American Journal of Physiology. Endocrinology and Metabolism. 280 (5): E745–E751. doi:10.1152/ajpendo.2001.280.5.e745. PMID11287357. S2CID24306481
Marette A (December 2003). “Molecular mechanisms of inflammation in obesity-linked insulin resistance”. International Journal of Obesity and Related Metabolic Disorders. 27 (Suppl 3): S46–S48. doi:10.1038/sj.ijo.0802500. PMID14704744. S2CID30693649.
“Reduce Abdominal Fat”. Archived from the original on 2011-09-28. Retrieved 2009-04-10. Estrogen causes fat to be stored around the pelvic region, hips, butt and thighs (pelvic region)
Researchers think that the lack of estrogen at menopause plays a role in driving our fat northward. See: Andrews M (2006-12-01). “A Matter of Fat”. Yahoo Health. Women’s Health. Archived from the original on 2007-03-15.
Manolopoulos KN, Karpe F, Frayn KN (June 2010). “Gluteofemoral body fat as a determinant of metabolic health”. International Journal of Obesity. 34 (6): 949–959. doi:10.1038/ijo.2009.286. PMID20065965. S2CID21052919.
Brodie D, Moscrip V, Hutcheon R (March 1998). “Body composition measurement: a review of hydrodensitometry, anthropometry, and impedance methods”. Nutrition. 14 (3): 296–310. doi:10.1016/S0899-9007(97)00474-7. PMID9583375.
Dhaliwal SS, Welborn TA (May 2009). “Central obesity and multivariable cardiovascular risk as assessed by the Framingham prediction scores”. The American Journal of Cardiology. 103 (10): 1403–1407. doi:10.1016/j.amjcard.2008.12.048. PMID19427436.
Atzmon G, Yang XM, Muzumdar R, Ma XH, Gabriely I, Barzilai N (November 2002). “Differential gene expression between visceral and subcutaneous fat depots”. Hormone and Metabolic Research. 34 (11–12): 622–628. doi:10.1055/s-2002-38250. PMID12660871. S2CID33960130.
Lemke, KA (June 2004). “Perioperative use of selective alpha-2 agonists and antagonists in small animals”. The Canadian Veterinary Journal. 45 (6): 475–80. PMC 548630. PMID 15283516.
Hedner T, Edgar B, Edvinsson L, Hedner J, Persson B, Pettersson AYohimbine pharmacokinetics and interaction with the sympathetic nervous system in normal volunteersEur J Clin Pharmacol.(1992)
Grossman E, Rosenthal T, Peleg E, Holmes C, Goldstein DSOral yohimbine increases blood pressure and sympathetic nervous outflow in hypertensive patientsJ Cardiovasc Pharmacol.(1993 Jul)
Berlan M, Galitzky J, Riviere D, Foureau M, Tran MA, Flores R, Louvet JP, Houin G, Lafontan MPlasma catecholamine levels and lipid mobilization induced by yohimbine in obese and non-obese womenInt J Obes.(1991 May)
Kaplan’s Essentials of Cardiac Anesthesia. Elsevier. 2018. doi:10.1016/c2012-0-06151-0. ISBN978-0-323-49798-5. Mechanisms of Action:ACE inhibitors act by inhibiting one of several proteases responsible for cleaving the decapeptide Ang I to form the octapeptide Ang II. Because ACE is also the enzyme that degrades bradykinin, ACE inhibitors increase circulating and tissue levels of bradykinin (Fig. 8.4).
Jandeleit-Dahm K, Cooper ME (Sep 2006). “Hypertension and diabetes: role of the renin–angiotensin system”. Endocrinol Metab Clin North Am. 35 (3): 469–90, vii. doi:10.1016/j.ecl.2006.06.007. PMID16959581.
Human Physiology, Silverthorn (Pearson Benjamin Cummings 2004)[page needed]
Weir M (1999). “The renin-angiotensin-aldosterone system: a specific target for hypertension management”. American Journal of Hypertension. 12 (4). Oxford University Press (OUP): 205–213. doi:10.1016/s0895-7061(99)00103-x. ISSN0895-7061. PMID10619573.
“Congestive Heart Failure”. The Lecturio Medical Concept Library. 7 August 2020. Retrieved 27 August 2021.
Kester M, Karpa KD, Vrana KE (2012). “Cardiovascular System”. Elsevier’s Integrated Review Pharmacology. Elsevier. pp. 125–151. doi:10.1016/b978-0-323-07445-2.00008-2. ISBN978-0-323-07445-2. ACE inhibitors also slow progression of kidney disease in patients with diabetic nephropathies. Renal benefits are probably a result of improved renal hemodynamics from decreased glomerular arteriolar resistance.
Long AN, Dagogo-Jack S. Comorbidities of diabetes and hypertension: mechanisms and approach to target organ protection. J Clin Hypertens (Greenwich) 2011; 13:244-251. http://doi:10.1111/j.1751-7176.2011.00434.x.
50. Palominos MM, Dünner DH, Wabitsch M, Rojas CV. 2015. Angiotensin II directly impairs adipogenic differentiation of human preadipose cells. Mol Cell Biochem 2015; 408: 115-122. http://doi:10.1007/s11010-015-2487-y.
51. Schling MM, Dünner NH, Wabitsch M, Rojas CV. Angiotensin II directly impairs adipogenic differentiation of human preadipose cells. Mol Cell Biochem 2015; 408:115-122. http://doi:10.1007/s11010-015-2487-y.
52. Brücher R, Cifuentes M, Acuña MJ, Albala C, Rojas CV. Larger anti-adipogenic effect of angiotensin II on omental preadipose cells of obese humans. Obesity 2007; 15:1643-1646. http://doi:10.1038/oby.2007.196.
53. Fuentes P, Acuña MJ, Cifuentes M, Rojas CV. The anti-adipogenic effect of angiotensin II on human preadipose cells involves ERK1,2 activation and PPARG phosphorylation. J Endocrinol 2010; 206:75-83. http:// doi:10.1677/JOE-10-0049.
Jones BH, Standridge MK, Moustaid N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology 1997; 138:1512-1519. http://doi:10.1210/endo.138.4.5038.
Townsend RR. The effects of angiotensin-II on lipolysis in humans. Metabolism 2001; 50:468-472. http://doi:10.1053/meta.2001.21021.
Weisinger RS, Begg DP, Jois M. Antagonists of the renin-angiotensin system and the prevention of obesity. Curr Opin Investig Drugs 2009; 10: 1069-1077.
Than A, Leow MK, Chen P. Control of adipogenesis by the autocrine interplays between angiotensin 1-7/Mas receptor and angiotensin II/AT1 receptor signaling pathways. J Biol Chem 2013; 288:15520-15531. http://doi:10.1074/jbc.M113.459792.
Sharma AM, Engeli S. The role of renin-angiotensin system blockade in the management of hypertension associated with the cardiometabolic syndrome. J Cardiometab Syndr 2006; 1:29-35. http://doi:10.1111/j.0197-3118.2006.05422.x.
Vertes V, Haynie R. Comparative pharmacokinetics of captopril, enalapril, and quinapril. Am J Cardiol. 1992;69:8C–16C. [PubMed] [Google Scholar]
Nakajima T, Yamada T, Setoguchi M. Prolonged inhibition of local angiotensin-converting enzyme after single or repeated treatment with quinapril in spontaneously hypertensive rats. J Cardiovasc Pharmacol. 1992;19:102–107. [PubMed] [Google Scholar]
Fukiyama F, Azuma J, Yoshida H, et al. A pharmacokinetic study of quinapril in normal Japanese men. J Clin Ther Med. 1993;9(Suppl 7):3–16. [Google Scholar]
Weishaar RE, Panek RL, Major TC, Simmerman J, Rapundalo ST, Taylor J, DG DG. Evidence for a functional tissue renin-angiotensin system in the rat mesentric vasculature and its involvement in regulation blood pressure. J Pharmacol Exp Ther. 1991;256:568–574. [PubMed] [Google Scholar]
Major TC, Overhiser RW, Taylor DG, Panek RL. Effect of quinapril, a new angiotensin-converting enzyme inhibitor, on vasoconstrictor activity in the isolated, perfused mesenteric vasculature of hypertensive rats. J Pharmacol Exp Ther. 1993;265:187–193. [PubMed] [Google Scholar]
Willenbrock R, Ozcelik C, Osterziel KJ, Dietz R. Angiotensin-converting enzyme inhibition, autonomic activity, and hemodynamics in patients with heart failure who perform isometric exercise. Am Heart J. 1996;131:999–1006. [PubMed] [Google Scholar]
Saitoh M, Miyakoda H, Kitamura H, Kinugawa T, Kotake H, Mashiba H. Effects of an angiotensin-converting enzyme inhibitor, alacepril, on cardiovascular and sympathetic nervous responses to mental stress in patients with essential hypertension. Intern Med. 1993;32:691–694. [PubMed] [Google Scholar]
Schling P, Mallow H, Trindl A, Loffler G. Evidence for a Local Renin Angiotensin System in Primary Cultured Human Preadipocytes. Int J Obes Relat Metab Disord (1999) 23(4):336–41. doi: 10.1038/sj.ijo.080082 PubMed Abstract | CrossRef Full Text | Google Scholar
Weisinger RS, Begg DP, Chen N, Jois M, Mathai ML, Sinclair AJ. The Problem of Obesity: Is There a Role for Antagonists of the Renin-Angiotensin System? Asia Pac J Clin Nutr (2007) 16 S1:359–67.Google Scholar
Takahashi N, Li F, Hua K, Deng J, Wang CH, Bowers RR, et al. Increased Energy Expenditure, Dietary Fat Wasting, and Resistance to Diet-Induced Obesity in Mice Lacking Renin. Cell Metab (2007) 6(6):506–12. doi: 10.1016/j.cmet.2007.10.01 PubMed Abstract | CrossRef Full Text | Google Scholar
Massiera F, Seydoux J, Geloen A, Quignard-Boulange A, Turban S, Saint-Marc P, et al. Angiotensinogen-Deficient Mice Exhibit Impairment of Diet-Induced Weight Gain With Alteration in Adipose Tissue Development and Increased Locomotor Activity. Endocrinology (2001) 142(12):5220–5. doi: 10.1210/endo.142.12.8556 PubMed Abstract | CrossRef Full Text | Google Scholar
Jayasooriya AP, Begg DP, Chen N, Mathai ML, Sinclair AJ, Wilkinson-Berka J, et al. Omega-3 Polyunsaturated Fatty Acid Supplementation Reduces Hypertension in TGR(mRen-2)27 Rats. Prostaglandins Leukot Essent Fatty Acids (2008) 78(1):67–72. doi: 10.1016/j.plefa.2007.11.001 PubMed Abstract | CrossRef Full Text | Google Scholar
De Blasi A, Cortellaro M, Costantini C. Enalapril in Essential Hypertension: A Comparative Study With Propranolol. Enalapril in Hypertension Study Group (Uk). Br J Clin Pharmacol (1984) 18(1):51–6. doi: 10.1111/j.1365-2125.1984.tb05021. PubMed Abstract | CrossRef Full Text | Google Scholar
Zorad S, Dou JT, Benicky J, Hutanu D, Tybitanclova K, Zhou J, et al. Long-Term Angiotensin II AT1 Receptor Inhibition Produces Adipose Tissue Hypotrophy Accompanied by Increased Expression of Adiponectin and Ppargamma. Eur J Pharmacol (2006) 552(1-3):112–22. doi: 10.1016/j.ejphar.2006.08.062 PubMed Abstract | CrossRef Full Text | Google Scholar
I capisaldi della gestione del peso sono gli interventi sullo stile di vita con il ruolo aggiuntivo di supporto dei farmaci anti-obesità e delle procedure bariatriche. Attualmente, i farmaci disponibili approvati per il trattamento dell’obesità agiscono sul bilancio energetico riducendo l’assunzione di cibo e il comportamento di ricompensa alimentare nel sistema nervoso centrale (ad esempio, sopprimendo l’appetito) o riducendo l’assorbimento dei grassi nell’intestino.[1,2,3] Finora non sono disponibili farmaci con un effetto diretto sull’aumento del dispendio energetico attraverso un’influenza sul tessuto adiposo [4,6]. Nell’uomo esistono due tipi di tessuto adiposo con funzioni fisiologiche distinte: il tessuto adiposo bianco (WAT), specializzato nell’immagazzinamento di trigliceridi in eccesso quando l’assunzione di energia supera il dispendio energetico, e il tessuto adiposo bruno (BAT) – con i relativi adipociti “beige”/”bruni” (derivati dal WAT) – che svolge un ruolo centrale nel metabolizzare il glucosio, gli acidi grassi e altre sostanze chimiche per produrre calore attraverso l’attivazione della proteina di disaccoppiamento 1 (UCP1) specifica del tessuto termogenico [4,7]. Alcuni dati suggeriscono che il BAT possa essere funzionale nell’uomo adulto [1]. I recettori β3-adrenergici (AR) sono espressi non solo nella vescica urinaria, ma anche sulla superficie degli adipociti bruni e bianchi [1]. I tessuti adiposi bruni e “beige”, contenenti cellule grasse termogeniche, possono essere attivati da agonisti dei recettori β3-adrenergici (β3-AR) [8]. È stato riportato che il Mirabegron, un agonista β3-AR umano selettivo, può stimolare il BAT e il processo di imbrunimento degli adipociti derivati dal WAT [9,10]. Il fatto che l’attivazione del BAT e degli adipociti “beige” possa aumentare il dispendio energetico rende i tessuti adiposi bruni e “beige” nuovi e promettenti bersagli per il trattamento dell’obesità [4,11].
Il ruolo del tessuto adiposo nella termogenesi e nei processi metabolici associati all’obesità:
Nell’uomo esistono due tipi principali di tessuto adiposo, che svolgono funzioni diverse: il tessuto adiposo bianco e il tessuto adiposo bruno. Oltre al WAT e al BAT, sono state distinte anche cellule adipose “brune”, definite cellule adipose “beige”. Esse derivano dal WAT, ma la loro funzione metabolica è simile a quella del BAT [4,7]. Il WAT è responsabile dell’immagazzinamento di energia sotto forma di trigliceridi, del rilascio di lipidi e della funzione di ghiandola endocrina, secernendo adipochine, come l’adiponectina e la leptina, per promuovere l’omeostasi metabolica [9,12]. Nell’obesità, gli adipociti bianchi si ipertrofizzano, seguiti da fibrosi, necrosi degli adipociti e infiltrazione di cellule immunitarie, che portano a infiammazione locale e sistemica, insulino-resistenza e disfunzione metabolica [9].
Cellule del BAT (colorate di marrone con anticorpi contro la proteina specifica del grasso bruno Ucp1) annidate tra le cellule del WAT grasso bianco.
Il BAT è stato descritto per la prima volta nel 1981 in finlandesi che lavoravano all’aperto e che erano stati esposti a basse temperature ambientali [6]. Il BAT metabolicamente attivo è stato identificato negli adulti mediante imaging PET/CT focalizzato principalmente sulla fossa sopraclavicolare, sull’area succlavia e sull’ascella, seguito dalle aree mediastiniche, paraspinali, perinefriche e sopradrenali [10,12]. Sebbene il BAT sia presente nell’uomo, la sua prevalenza diminuisce con l’età e nelle persone in sovrappeso o obese rispetto ai soggetti magri [6,9,13,14]. Gli anelli mancanti nel trattamento dell’obesità sono i farmaci che possono aumentare la quantità o l’attività del BAT. È stato riportato che il volume del BAT può essere aumentato dopo la chirurgia bariatrica [12]. Il BAT è il principale organo termogenico dei mammiferi, con lo scopo di aumentare il dispendio energetico in risposta al freddo o ad altre stimolazioni nervose simpatiche, rilasciando noradrenalina dai terminali nervosi per attivare i recettori β3-adrenergici attraverso il processo definito termogenesi senza brividi [2,10,11,12,13]. La capacità termogenica del BAT è stata stimata in circa 500 W/kg [6]. Gli adipociti del BAT sono arricchiti di mitocondri (i loro livelli sono più alti di quelli del WAT), nei quali la proteina di disaccoppiamento 1 (UCP1) è altamente espressa. La UCP1 dissipa l’energia in eccesso sotto forma di calore in un processo noto come termogenesi [2,15]. L’attivazione adrenergica della lipolisi stimola l’attività termogenica della UCP1 [2,10]. L’attivazione dell’UCP1 sulla membrana mitocondriale interna disaccoppia la respirazione mitocondriale, separando il trasporto di elettroni dalla produzione di ATP per ossidare il substrato e generare calore [4,8,16]. Gli acidi grassi a catena lunga, generati dai pool lipidici intracellulari, sono trasportati ai mitocondri attraverso la carnitina palmitoiltransferasi 1 (CPT1) e utilizzati come fonte di carburante dagli adipociti bruni per produrre calore. Inoltre, è stato proposto che gli acidi grassi liberi agiscano come attivatori allosterici di UCP1. Oltre agli acidi grassi, anche il glucosio circolante può essere utilizzato dal BAT attivo per alimentare la termogenesi [2,10,17]. In sintesi, il BAT consuma glucosio e lipidi per generare calore attraverso la respirazione disaccoppiata mediata da UCP1, con conseguente miglioramento dell’omeostasi glucidica e lipidica [9,13,18].
Struttura della proteina disaccoppiante umana UCP1
Gli adipociti termogenici umani possono originare da due lignaggi distinti, non solo da adipociti bruni costitutivi ma anche da cellule “beige” reclutabili, definite adipociti “bruni” o “bruno-simili” [5]. Gli adipociti “beige” sono localizzati prevalentemente nei depositi di WAT [16]. Le cellule adipose del WAT possono essere convertite in adipociti “beige” termogenici in un processo chiamato “browning” o “beiging” [12]. Da un lato, è stato dimostrato un sostanziale “beiging” del WAT sottocutaneo umano in alcuni disturbi, come la cachessia da cancro, le ustioni e le condizioni con alti livelli di catecolamine, ad esempio il feocromocitoma [8,13,14]. I pazienti con tumori che secernono catecolamine hanno anche più tessuto adiposo bruno rispetto alla maggior parte delle persone [19]. D’altra parte, gli adipociti “bruni” possono essere attivati con l’induzione dell’espressione di UCP1 da parte di stimoli ambientali, come l’esposizione al freddo e agli agonisti β-adrenergici, mediata dalla via di segnalazione p38-MAPK [9,12,14,16]. La risposta “beiging” dei soggetti obesi al freddo è simile a quella dei soggetti magri [14]. Sebbene queste cellule differiscano dagli adipociti bruni convenzionali – in quanto si sviluppano da una cellula precursore di adipociti bianchi e non da una cellula precursore di adipociti bruni, simile agli adipociti bruni classici nel BAT – gli adipociti “beige” possiedono goccioline lipidiche multiloculari, un gran numero di mitocondri e marcatori unici di espressione genica del grasso bruno, come UCP1, aumentando la capacità del tessuto di ossidazione del carburante e il dispendio energetico [4,16,18]. Inoltre, è stato dimostrato che il “beiging” è associato a una riduzione della fibrosi del tessuto adiposo e della disfunzione adiposa. Questi risultati suggeriscono che l’induzione del tessuto adiposo “beige” può migliorare l’omeostasi metabolica aumentando la capacità del WAT sottocutaneo di funzionare come serbatoio metabolico per il glucosio e i lipidi o riducendo la disfunzione del WAT che si verifica con l’obesità [13]. Così, oltre alla termogenesi e al dispendio energetico, i tessuti adiposi bruni e “beige” sono associati a un miglioramento dell’omeostasi del glucosio e dei lipidi, nonché a una maggiore sensibilità all’insulina nell’uomo e nel topo [14].
Distribuzione del BAT nei neonati, nelle donne e negli uomini. Il BAT è immagazzinato in un deposito interscapolare separato nei neonati che perdono il loro tessuto adiposo bruno con l’avanzare dell’età. Negli esseri umani adulti, la maggior parte degli adipociti bruni si trova nei depositi di BAT sopraclavicolari nella regione del collo. Quantità minori di BAT si trovano nell’aorta, nelle vertebre, nelle aree ascellari e renali. C’è una distribuzione simile del tessuto adiposo bruno sia nelle donne che negli uomini. Tuttavia, le donne hanno una maggiore quantità di massa e attività di BAT.
Considerando il fatto che nelle persone obese adulte c’è meno BAT rispetto ai soggetti magri, il WAT in eccesso, che può essere stimolante e in fase di “beigezzazione/imbrunimento”, può svolgere un ruolo aggiuntivo rispetto al BAT nei processi metabolici [13]. Pertanto, il tessuto adiposo bruno e il tessuto adiposo “beige” sono stati riconosciuti come regolatori critici del metabolismo e del dispendio energetico dell’intero corpo e sono considerati bersagli promettenti per la terapia anti-obesità [2,12,15].
L’Irisina è un ormone sintetizzato in grande quantità dal tessuto muscolare umano durante le attività sportive. La molecola è in grado di operare il meccanismo molecolare detto “browning” [“imbrunimento”], ovvero di conversione del WAT in BAT.
Il grasso bruno, il grasso “beige” e i β3-adrenocettori nel contesto dell’obesità:
Struttura del recettore β3-adrenergico
La famiglia dei recettori β-adrenergici (AR) umani è composta dai recettori β1, β2 e β3, in cui il β1-AR è altamente espresso in tutto il sistema cardiovascolare, il β2-AR si trova nelle vie aeree polmonari, in tutta la vascolarizzazione e nel muscolo scheletrico e l’espressione del β3-AR è limitata soprattutto alla vescica urinaria e alla cistifellea, oltre che al BAT e al WAT [4,10]. Il β3-AR umano, identificato nel 1989, è un recettore a 7 membrane, con una coda N-terminale extracellulare e una coda C-terminale intracellulare, composta da 408 aminoacidi. Si accoppia principalmente a Gs per attivare l’adenilato ciclasi, con conseguente aumento dei livelli intracellulari di cAMP, sebbene sia stato riportato un accoppiamento promiscuo con altri effettori, come Gi [4,16].
La β3-AR svolge un ruolo critico nel tessuto adiposo, nella regolazione della termogenesi, della glicolisi e della lipolisi [16]. Studi sugli animali hanno dimostrato che la stimolazione cronica del BAT porta a un miglioramento della tolleranza al glucosio e della sensibilità all’insulina e a una riduzione dell’obesità, oltre che al rilascio di adipochine che regolano beneficamente il metabolismo [1,8,10,12,20]. Inoltre, l’attivazione β3-AR-mediata del WAT può aumentare la secrezione insulinica delle cellule β pancreatiche [5]. È stato anche riportato che una parte significativa della termogenesi non da brivido ha luogo nel tessuto adiposo bruno ed è mediata principalmente dal β3-adrenocettore [19]. Nei topi alimentati con dieta a base di chow e ad alto contenuto di grassi, il trapianto di BAT ha ridotto il peso corporeo, aumentato il metabolismo del glucosio e la sensibilità all’insulina e incrementato l’assorbimento di glucosio nel BAT e nel WAT [15].
Oltre alla funzione metabolica, la β3-AR svolge un ruolo nel cervello, essendo coinvolta nei processi di memoria, apprendimento e regolazione dell’appetito, nel tratto gastrointestinale, dove partecipa alla regolazione della motilità, e nel sistema genitourinario, dove svolge un ruolo nella regolazione della funzione vescicale [16].
Durante la termogenesi, i β3-adrenocettori aumentano il dispendio energetico, che può portare alla perdita di grasso, in risposta alla stimolazione simpatica [19]. È dimostrato che la stimolazione cronica dell’attività nervosa simpatica e dei β3-AR può attivare il BAT [6]. È stato dimostrato che l’esposizione al freddo stimola il sistema nervoso simpatico a rilasciare noradrenalina dalle terminazioni nervose simpatiche per attivare i β-AR sulle membrane delle cellule del BAT, promuovendo la termogenesi. In questo modo, il BAT umano è in grado di avviare la termogenesi attraverso il consumo di acidi grassi e glucosio e, successivamente, di generare calore [1,3,8]. Inoltre, l’attivazione dei β3-ARs da parte dell’esposizione al freddo o di agenti farmacologici induce un programma di “beiging” nel WAT [18]. Un modo per aumentare la quantità effettiva di tessuto adiposo bruno può essere quello di somministrare l’agonista β3-adrenoccettore in modo cronico [19]. Una singola dose di agonista dei β3-adrenocettori può almeno raddoppiare il dispendio energetico in un modello murino a circa 21 °C [19].
Struttura del gene ADRB3
Il ruolo dei β3-AR nel metabolismo energetico umano è supportato da studi clinici che riportano associazioni tra polimorfismi specifici nel gene umano ADRB3 (il gene che codifica i β3-AR) e tassi più elevati di obesità, insulino-resistenza e diabete [10]. Inoltre, le mutazioni nel gene ADRB3 sono state correlate all’insulino-resistenza, all’aumento del rischio di obesità e diabete e alla malattia del fegato grasso non alcolico negli individui obesi [10]. I dati indicano che il silenziamento di ADRB3 negli adipociti umani “marroni”/”beige” altera il macchinario termogenico cellulare e causa una riduzione dei livelli di espressione dei geni associati al metabolismo degli acidi grassi, alla massa mitocondriale e alla termogenesi, senza compromettere il fenotipo “marrone”/”beige” [10].
Attività agonista dei β3-AR del Mirabegron:
Struttura molecolare del Mirabegron
Mirabegron è una nuova generazione di agonisti dei β3-adrenocettori con una buona biodisponibilità [21]. Gli effetti dell’agonista selettivo dei β3-AR mirabegron sul rilassamento della vescica sono stati scoperti nel 2007. Per la prima volta, la selettività β3 del mirabegron (YM-178) nel contesto della funzione vescicale è stata descritta da Takasu et al. [22]. YM-178 ha aumentato l’accumulo di AMP ciclico in cellule ovariche di criceto cinese che esprimono il β3-adrenocettore umano. Mirabegron ha dimostrato valori di EC50 nanomolari contro il β3-AR umano in saggi biochimici, con una potente selettività rispetto ai β1- e β2-AR [22]. Studi in vivo hanno dimostrato che la somministrazione di mirabegron ha ridotto la pressione intravescicale e le contrazioni spontanee della vescica in modo dose-dipendente [23]. Mirabegron è stato approvato dalla Food and Drug Administration (FDA) statunitense nel 2012 come nuovo tipo di trattamento farmacologico per la vescica iperattiva (OAB) [6,21,24]. Cinquanta milligrammi di mirabegron è la dose raccomandata a tutti i pazienti con OAB [24]. Il farmaco è generalmente ben tollerato e gli effetti collaterali più comuni includono ipertensione, rinofaringite e infezione del tratto urinario [6].
La selettività β3 di Mirabegron è stata confermata in molti studi con l’uso di linee cellulari che esprimono il β3-adrenocettore sia animale che umano [22,23,25]. Mirabegron ha mostrato una selettività per il β3-AR umano superiore di oltre 400 volte rispetto al β1-AR o al β2-AR umano [26]. Ad esempio, Brucker et al. [27] hanno utilizzato cellule di ovaio di criceto cinese (CHO)-K1, cellule di rene embrionale umano 293 esprimenti stabilmente recettori β1-, β2- o β3-adrenergici umani e recettori α1D- e α2B-adrenergici umani per valutare la selettività di mirabegron. A una concentrazione di 10 μM, l’attività β3-adrenergica rispetto all’isoproterenolo (agonista β-adrenergico completo) era dell’88% per mirabegron. A sua volta, l’attività β1- e β2-adrenergica di mirabegron era rispettivamente del 3% e del 15% [27]. In questo studio mirabegron non ha soddisfatto il criterio di significatività per l’inibizione dei recettori α1D- o α2B-adrenergici [27]. Tuttavia, alcuni studi hanno indicato che mirabegron potrebbe svolgere un ruolo come antagonista degli α1-adrenergici [28,29]. Alexandre et al. [28] hanno ipotizzato che mirabegron rilassasse la muscolatura liscia uretrale nei topi attraverso un duplice meccanismo che coinvolge l’attivazione dei β3-adrenocettori e il blocco degli α1-adrenocettori. In un altro studio, mirabegron ha indotto una vasorilassazione endotelio-indipendente nelle arterie del tessuto adiposo viscerale attraverso l’antagonismo degli α1-adrenocettori. Questa azione ha suggerito che mirabegron potrebbe migliorare efficacemente la perfusione del tessuto adiposo viscerale, favorendo così un sano rimodellamento del tessuto adiposo e prevenendo alcune delle conseguenze cardiometaboliche indesiderate dell’obesità e dell’invecchiamento [29]. Resta ancora difficile stabilire in che misura l’antagonismo degli α1-adrenocettori possa contribuire agli effetti clinici di mirabegron [28,29].
I cambiamenti metabolici benefici causati dal trattamento cronico con mirabegron potrebbero derivare dalla stimolazione della β3-AR nel BAT e nel WAT umani [5,12,18]. È stato suggerito che mirabegron potrebbe migliorare le malattie metaboliche legate all’obesità aumentando la termogenesi del BAT, la lipolisi del WAT e la stimolazione del processo di “brunimento” degli adipociti derivati dal WAT [4,5,9,10]. Il trattamento acuto con mirabegron ha aumentato il dispendio energetico [10,15]. Dopo il silenziamento dell’espressione dei β3-AR, il mirabegron non è stato in grado di stimolare la lipolisi e la termogenesi del BAT [10]. Molti studi hanno dimostrato che il trattamento con mirabegron ha aumentato l’assorbimento del glucosio negli adipociti bruni e “beige”, ha migliorato l’omeostasi del glucosio e ha aumentato la sensibilità all’insulina e la funzione delle cellule β [1,9]. Inoltre, è stato dimostrato che il trattamento cronico con agonisti β3-AR nell’uomo può rilasciare adipochine benefiche [1]. Il modo in cui mirabegron migliora il metabolismo del glucosio non è stato finora chiarito [5]. Tuttavia, sono stati ipotizzati alcuni meccanismi. In primo luogo, mirabegron stimola la secrezione di adiponectina, nota adipochina derivata dal WAT e associata a una maggiore sensibilità all’insulina nel muscolo scheletrico e nel fegato. In secondo luogo, mirabegron aumenta la concentrazione di polipeptide inibitore gastrico (GIP), l’incretina collegata alla secrezione di insulina. Infine, il meccanismo di mirabegron potrebbe coinvolgere le stesse cellule β [5].
L’agonista dei recettori β3-adrenergici è un ottimo candidato per il trattamento dell’obesità, poiché l’isoforma β3 è espressa esclusivamente negli adipociti e l’azione su altri tipi di cellule, come i cardiomiociti e le cellule muscolari lisce, attraverso le altre isoforme β – β1 e β2 – è minima e dose-dipendente [11]. Pertanto, come agonista β3-AR, Mirabegron attiverebbe la termogenesi nel tessuto adiposo, stimolando l’ossidazione dei lipidi e il consumo di glucosio per produrre calore, senza causare gravi effetti collaterali cardiovascolari [13].
Mirabegron come agente antiobesità negli studi sperimentali:
Adipocita del BAT
Il trattamento dei roditori con agonisti β3-AR ha attivato il BAT, con conseguente aumento del dispendio energetico, perdita di peso e miglioramento del metabolismo del glucosio e dei lipidi. Inoltre, ha ripristinato l’equilibrio NO/redox, migliorato la funzione endoteliale e, quindi, esercitato effetti protettivi vascolari [4,6,13,17]. L’aumento dell’attività del BAT ha impedito lo sviluppo e la gravità dell’obesità e del diabete di tipo 2, mentre i topi privi di BAT erano inclini all’obesità [16]. È stato riportato che una riduzione della massa del BAT nei topi indotta da un transgene produce obesità e che questi topi presentano un’ulteriore maggiore suscettibilità all’obesità a causa di diete obesitogene [8,30,31].
Adipocita “Beige”
Come si è detto, il Mirabegron può essere efficace come attivatore del BAT, stimolatore delle cellule “beige” e controllore dell’omeostasi metabolica. L’influenza benefica di mirabegron sul metabolismo è stata confermata da studi in vitro e in vivo [2,4,15,18]. Nello studio condotto da Dehvari et al. [15], sono stati riportati gli effetti di mirabegron negli adipociti bruni, bianchi e “beige” in vitro e i suoi effetti sull’utilizzo del glucosio e sulla termogenesi in vivo. È stato dimostrato che mirabegron aumenta l’assorbimento di glucosio e la glicolisi negli adipociti bruni di topo in vitro e promuove l’assorbimento di glucosio nel BAT in vivo. Il mirabegron ha aumentato i livelli di cAMP e l’mRNA di UCP1, con conseguente aumento del consumo di ossigeno mediato da UCP1, nonché l’assorbimento di glucosio e la glicolisi cellulare negli adipociti bruni e “beige” (tale azione è mancata nelle colture cellulari primarie di adipociti bruni provenienti da topi knockout per il β3-adrenocettore), mentre questi effetti erano assenti o ridotti negli adipociti bianchi. In vivo, mirabegron ha aumentato il consumo di ossigeno nell’intero corpo e l’assorbimento di glucosio nel tessuto adiposo bruno e bianco inguinale e ha migliorato la tolleranza al glucosio. Nei topi knockout per il β3-adrenorecettore, mirabegron non è riuscito a indurre l’assorbimento di glucosio nel tessuto adiposo, né ad aumentare il consumo di ossigeno corporeo, il che dimostra che la segnalazione del β3-adrenorecettore è una via principale delle azioni metaboliche di mirabegron [15]. Analogamente a Dehvari et al. [15], Hao et al. [4] hanno studiato gli effetti anti-obesità di mirabegron utilizzando modelli in vitro e in vivo. In entrambe le linee cellulari – preadipociti bruni di topo e preadipociti bianchi 3T3-L1 – mirabegron ha stimolato l’espressione di UCP1. I topi trattati con mirabegron, alimentati con una dieta ad alto contenuto di grassi, presentavano una riduzione del peso corporeo e dell’adiposità, nonché un miglioramento della tolleranza al glucosio e della sensibilità all’insulina. Le goccioline lipidiche nel BAT dei topi trattati con mirabegron erano meno numerose e di dimensioni inferiori rispetto ai controlli. La colorazione H&E e l’immunoistochimica hanno indicato che mirabegron ha aumentato l’abbondanza di cellule “beige” nel WAT [4]. Si è concluso che mirabegron ha aumentato l’espressione di UCP1 e ha promosso la “brunitura” del WAT, che è stata accompagnata da un miglioramento della tolleranza al glucosio, della sensibilità all’insulina e della prevenzione dell’obesità indotta da una dieta ad alto contenuto di grassi [4]. In un altro studio su animali, Valgas da Silva et al. [18] hanno riferito che un trattamento di 2 settimane con mirabegron ha ridotto l’infiammazione, migliorato il metabolismo, impedito l’accumulo di grasso ectopico nel BAT e nel fegato e diminuito l’insulino-resistenza nei topi obesi (riduzione dell’indice HOMA e dei livelli di insulina). Mirabegron ha aumentato l’espressione di UCP1 nel BAT e il dispendio energetico, oltre a ridurre l’adiposità nei topi obesi. Inoltre, mirabegron ha ridotto i livelli circolanti di acidi grassi liberi, glicerolo e TNF-α. È noto che l’aumento dei livelli di FFA circolanti causa insulino-resistenza negli organi bersaglio dell’insulina ed è emerso come uno dei principali collegamenti tra l’obesità e lo sviluppo della sindrome metabolica. È noto anche che il TNF-α ha un effetto lipolitico, che determina un aumento dei livelli di FFA e glicerolo in circolo, contribuendo all’insulino-resistenza. Tuttavia, a differenza dello studio condotto da Dehvari et al. non sono stati riscontrati cambiamenti nel WAT inguinale: il mirabegron non ha indotto il “beiging” del WAT inguinale dei topi obesi. Inoltre, l’obesità indotta dalla dieta ha aumentato significativamente i depositi lipidici nel fegato e nel BAT, ma mirabegron ha parzialmente invertito questi cambiamenti, il che potrebbe indicare un ruolo protettivo di mirabegron nello sviluppo della steatosi epatica e dell’insulino-resistenza [18]. La conferma che mirabegron può essere utile come agente anti-obesità è stata trovata anche nello studio di Hao et al. [4]. È stato dimostrato che mirabegron provoca un aumento di 14 volte dell’espressione genica di UCP1 e può determinare una perdita di peso del 12% e una riduzione dell’adiposità nei topi obesi rispetto all’attività fisica.
Struttura molecolare della Metformina
La terapia combinata, composta da Mirabegron e Metformina, è stata verificata nel modello murino di prevenzione e nel modello murino di trattamento dell’obesità [2]. La metformina, un derivato della biguanide, è uno dei farmaci più comunemente utilizzati per il trattamento del diabete di tipo 2. Inibisce il complesso mitocondriale I, vitale per il trattamento dell’obesità. Inibisce il complesso mitocondriale I, vitale per il trasporto di elettroni, che porta all’attivazione dell’AMPK (proteina chinasi attivata dall’adenosina 5′-monofosfato). Di conseguenza, la produzione di ATP (adenosina trifosfato) diminuisce e la concentrazione intracellulare di ADP (adenosina difosfato) aumenta. Di conseguenza, i livelli cellulari di AMP (adenosina monofosfato) aumentano, attivando infine l’AMPK. L’AMPK è un regolatore chiave di numerose vie metaboliche, tra cui il metabolismo del glucosio e dei lipidi e l’omeostasi energetica. La metformina svolge anche un ruolo importante inibendo la segnalazione dei recettori dell’insulina e dell’IGF, con conseguenti cambiamenti nell’omeostasi metabolica [32]. Zhao et al. [2] hanno indicato che questa terapia complessa potrebbe essere un approccio promettente per la prevenzione e il trattamento dell’obesità, agendo contemporaneamente sull’assunzione e sul dispendio energetico, senza effetti collaterali sulla funzione cardiovascolare. Nel modello di prevenzione, metformina e mirabegron hanno provocato un’ulteriore riduzione del 12% e del 14% dell’aumento di peso corporeo indotto da una dieta ad alto contenuto di grassi, rispetto a metformina o mirabegron da soli, rispettivamente. Nel modello di trattamento, metformina e mirabegron hanno promosso in modo additivo una perdita di peso corporeo del 17% nei topi obesi indotti dalla dieta, superiore del 13% e del 6% rispetto a metformina e mirabegron da soli, rispettivamente. La terapia combinata ha avuto un effetto additivo sulla perdita di peso nei topi, associato a una significativa perdita di grasso, soprattutto nel WAT sottocutaneo [2]. I ricercatori hanno suggerito che l’effetto additivo di metformina e mirabegron sull’aumento del dispendio energetico abbia contribuito in modo determinante alla riduzione del peso corporeo e della massa grassa nei topi [2]. La terapia con metformina e mirabegron ha avuto un effetto additivo sulla termogenesi del BAT e sulla doratura del WAT sottocutaneo. La terapia combinata ha aumentato significativamente l’espressione di UCP1 nel BAT e nel WAT sottocutaneo [2]. Inoltre, metformina e mirabegron hanno migliorato la tolleranza al glucosio e la sensibilità all’insulina, e l’effetto era indipendente dall’assunzione di cibo. Tuttavia, la co-somministrazione di metformina e mirabegron non ha migliorato l’omeostasi del glucosio nei topi in misura maggiore rispetto alla metformina o al mirabegron da soli [2].
Un diagramma che riassume gli effetti combinati di Metformina (Met)/Mirabegron (Mir) sull’obesità nei modelli di prevenzione e trattamento. (A) Nel modello di prevenzione in cui una dieta ricca di grassi (HFD) e farmaci venivano somministrati simultaneamente, il trattamento con Met/Mir ha ridotto l’aumento di peso in modo additivo. Ciò è dovuto principalmente a un miglioramento della spesa energetica (EE) che era accompagnato da un’espressione sovraregolata di marcatori critici nella lipolisi, nell’ossidazione degli acidi grassi e nella termogenesi nel tessuto adiposo bruno (BAT). (B) Nel modello di trattamento, è stato prima stabilito un fenotipo di obesità indotta dalla dieta (DIO), seguito da 5 settimane di trattamenti terapeutici con Met e/o Mir. Il trattamento con Met/Mir ha causato una marcata perdita di peso, derivante dall’aumento di EE
E’ interessante notare che la p-Sinefrina, una agonista selettivo dei β3-Adrenocettoiri di origine naturale, ha mostrato in studi su animali effetti positivi sull’imbrunimento del WAT, sopprimendo così l’obesità e la steatosi epatica.
Mirabegron come farmaco antiobesità: i dati degli studi sull’uomo:
Oltre agli studi sperimentali, esistono numerosi studi clinici in cui è stata dimostrata l’influenza di mirabegron sull’attività del BAT e sulla massa corporea. Gli autori hanno riferito che mirabegron ha portato a un aumento dell’attività del BAT e del dispendio energetico a riposo [1,3,5,10,17,21]. Prove preliminari suggeriscono che gli effetti del mirabegron sul metabolismo del glucosio, sul colesterolo HDL e sugli acidi biliari assomigliano a quelli ottenuti con un lieve esercizio fisico [1,5]. Nel primo gruppo di studi sono state testate soprattutto dosi elevate di mirabegron (100 mg, 150 mg o 200 mg) [1,3,5,17,21].
Cypess et al. [1] hanno usato, per la prima volta, Mirabegron per studiare il BAT umano e hanno confrontato la sua azione in un grado che corrispondeva alle risposte all’esposizione al freddo. La somministrazione di 200mg al giorno di Mirabegron orale per 12 settimane a 12 uomini sani è stata associata a una maggiore attività del BAT (misurata tramite tomografia a emissione di positroni 18F-fluorodesossiglucosio combinata con tomografia computerizzata) e all’aumento del tasso metabolico a riposo di 203 ± 40 kcal/die, rispetto agli individui che hanno ricevuto il placebo. È stato ipotizzato che la perdita di peso calcolata, associata al dispendio energetico, dovrebbe raggiungere i 5 kg nel primo anno e i 10 kg entro la fine dei 3 anni [1]. In questo studio, il dosaggio di 200 mg di mirabegron, una dose molto più alta di quelle attualmente approvate per ridurre i sintomi della vescica iperattiva, è stato generalmente ben tollerato, anche dopo 12 settimane di somministrazione orale giornaliera [1]. L’effetto collaterale più comune era la tachicardia [1].
L’alta dose di Mirabegron [100mg al giorno] è stata testata da O’Mara et al. durante un programma di terapia di 4 settimane su 14 donne sane di varie etnie [5]. Nell’endpoint primario, i ricercatori hanno riferito che la terapia cronica con Mirabegron ha aumentato il volume del BAT e l’attività metabolica, misurati tramite PET/CT con 18F-fluorodesossiglucosio [5]. Inoltre, le donne che avevano avuto principalmente meno BAT hanno finalmente raggiunto un aumento maggiore del volume e dell’attività del BAT dopo il trattamento [5]. Gli endpoint secondari hanno rivelato che la spesa energetica a riposo dell’intero corpo era più alta dopo il trattamento con mirabegron; tuttavia, non sono state riscontrate modifiche nel peso corporeo o nella composizione. Questi risultati dovrebbero essere associati a un intervallo di BMI ristretto e alla partecipazione di donne non obese. Inoltre, è stato riscontrato che la terapia con Mirabegron aumenta i biomarcatori delle lipoproteine come HDL e apolipoproteina A1, apolipoproteina E e peptide inibitorio gastrico (GIP), nonché i livelli di adiponectina, adipochina antidiabetica e antinfiammatoria. Dopo il trattamento con mirabegron, è stata osservata una riduzione del rapporto ApoB100/ApoA1, un biomarcatore del rischio cardiovascolare. Infine, dopo il trattamento cronico con mirabegron, un test di tolleranza al glucosio per via endovenosa ha rivelato una maggiore sensibilità all’insulina, efficacia del glucosio e secrezione di insulina [5]. Tuttavia, il cambiamento nella valutazione del modello omeostatico della resistenza all’insulina (HOMA-IR), una misura della resistenza all’insulina, non è stato significativo dopo il trattamento cronico con mirabegron. Gli autori hanno suggerito che la ragione principale dovrebbe essere il livello HOMA-IR quasi normale all’inizio dello studio [5]. Come è una preoccupazione comune nel trattamento cronico con agonisti adrenergici, 100mg di Mirabegron hanno portato a una variazione diurna della frequenza cardiaca tale che Mirabegron l’ha aumentata di più durante la notte rispetto a quando i soggetti erano svegli e in movimento. D’altra parte, il trattamento con Mirabegron non ha avuto alcun effetto sulla tolleranza all’esercizio [5].
Loh et al. [21] hanno riportato l’efficacia di varie dosi singole di mirabegron (50, 100, 150 e 200 mg) in un gruppo di 17 individui sani (11 uomini, 6 donne) che hanno assunto il farmaco in quattro giorni separati, con 3-14 giorni di wash-out tra ogni dose. Hanno riferito che la spesa energetica (misurata tramite calorimetria indiretta) è aumentata significativamente dopo le dosi da 100 mg e 200 mg e ha mostrato una tendenza all’aumento dopo le dosi da 150 mg, ma non era significativamente diversa dal basale in risposta a 50 mg di mirabegron. La temperatura cutanea sopraclaveare (come indicatore surrogato dell’attività BAT), è aumentata dopo le dosi di mirabegron da 50 mg, 100 mg e 150 mg, ma non era significativamente diversa dal basale in risposta a 200 mg. Considerando gli effetti collaterali, il cambiamento nella pressione sanguigna sistolica è stato significativo dopo le dosi da 150 mg e 200 mg rispetto alla dose da 50 mg e alla dose da 100 mg. Tuttavia, non c’era alcuna differenza nella pressione sanguigna diastolica tra le dosi da 50 mg, 100 mg, 150 mg e 200 mg. Il cambiamento nella frequenza cardiaca è stato maggiore dopo 200 mg rispetto alle dosi rimanenti. Hanno concluso che una dose da 100 mg di mirabegron può essere efficace per aumentare il dispendio energetico e la temperatura cutanea sopraclaveare in modo specifico per il recettore β3-adrenergico, senza gli aumenti significativi della pressione sanguigna o della frequenza cardiaca osservati a dosi più elevate [21].
Baskin et al. [17] hanno studiato le implicazioni cliniche del mirabegron in 12 uomini sani e magri a cui è stata somministrata la dose approvata di 50 mg e una dose elevata di 200 mg. Si è verificato un aumento più che proporzionale alla dose nell’attività metabolica del BAT (misurata tramite PET/CT). Rispetto al placebo, 50 mg di mirabegron hanno aumentato l’attività del BAT nella maggior parte dei soggetti. Tuttavia, l’attivazione del BAT con 50 mg è stata significativamente inferiore rispetto a quella con 200 mg. Solo la dose da 200 mg ha aumentato la spesa energetica a riposo (5,8%). La stimolazione cardiovascolare è stata coerente con studi precedenti, poiché 200 mg di mirabegron hanno aumentato sia la frequenza cardiaca che la pressione sanguigna.
Uno studio randomizzato, in doppio cieco, cross-over costituito da tre interventi (esposizione al freddo a breve termine (~2 h), mirabegron (dose singola da 200 mg) e placebo) in un gruppo di 10 uomini magri olandesi sud asiatici e 10 uomini magri europei, condotto da Nahon et al. [3], ha rivelato che l’esposizione al freddo e il mirabegron hanno indotto effetti metabolici benefici, tra cui un aumento della spesa energetica a riposo (misurata mediante calorimetria indiretta), livelli di acidi grassi liberi nel siero e ossidazione dei lipidi. Il mirabegron ha aumentato la frequenza cardiaca sia nei sud asiatici (+10 battiti/min) che nei caucasici bianchi (+7 battiti/min), mentre la pressione sanguigna sistolica e diastolica non sono cambiate in modo significativo [3]. È stato osservato che una singola dose di mirabegron ha aumentato i livelli di insulina nel siero senza influenzare i livelli di glucosio. Il mirabegron può stimolare il rilascio di insulina direttamente agendo sul β3-AR del pancreas o indirettamente attraverso un aumento degli FFA che possono stimolare il pancreas a rilasciare insulina [3].
L’azione dose-dipendente del mirabegron sul tessuto adiposo, inclusa l’influenza sull’attività BAT e sul dispendio energetico, può essere analoga all’effetto del mirabegron sulla vescica urinaria. L’attivazione dei recettori β3-adrenergici con mirabegron ha determinato risposte dei recettori β3-adrenergici dipendenti dalla concentrazione [27]. Per quanto riguarda la funzione della vescica, negli studi in vivo, la somministrazione di mirabegron ha ridotto la pressione intravescicolare e le contrazioni vescicali spontanee in modo dose-dipendente [23].
È stato riportato che dosi elevate di mirabegron (in particolare 200 mg al giorno), molto più elevate di quelle approvate dalla FDA per l’iperattività della vescica (50 mg al giorno), possono essere associate a effetti collaterali cardiovascolari come mal di testa, tachicardia e pressione sanguigna elevata (per lo più solo pressione sanguigna sistolica) [1,3,5,17,21]. L’aumento della pressione sanguigna sistolica può raggiungere ~10 mm Hg alla dose di 200 mg al giorno [21]. Questo è il risultato della perdita di selettività per il β3-adrenocettore a questa dose, tale che mirabegron attiva indirettamente i β1-adrenocettori che sono ampiamente espressi in vari organi, in particolare il sistema cardiovascolare. Questo meccanismo coinvolge l’assorbimento del trasportatore di noradrenalina del mirabegron nei terminali nervosi simpatici cardiaci, causando successivamente un rilascio di noradrenalina, che attiva i β1-adrenocettori [21]. Tuttavia, il trattamento con mirabegron non ha avuto effetti sulla tolleranza all’esercizio [5]. L’attivazione dei β1-adrenocettori può essere attenuata dalla co-somministrazione di propranololo o bisoprololo [16]. D’altro canto, gli studi clinici hanno rivelato che dosi di mirabegron fino a 100 mg al giorno per almeno 12 mesi hanno mostrato un buon profilo di sicurezza e non hanno determinato un aumento dell’incidenza di tachicardia, pressione sanguigna, alterazioni dell’ECG o eventi cardiovascolari [21]. Dosi terapeutiche inferiori (50 mg) nei pazienti con OAB hanno determinato piccole variazioni della frequenza cardiaca (1 battito al minuto) e della pressione sanguigna (1 mm Hg o meno). Considerando gli effetti collaterali cardiovascolari, il mirabegron non è raccomandato nei pazienti con grave ipertensione incontrollata (pressione sanguigna sistolica ≥ 180 mm Hg e/o pressione sanguigna diastolica ≥ 110 mm Hg) [16].
Nel secondo gruppo di studi, condotto da Finlin et al. [9,13,14], è stata testata una bassa dose di mirabegron, una che è stata approvata per il trattamento dell’OAB. In un gruppo di 13 pazienti obesi di mezza età, 50 mg di mirabegron al giorno durante una terapia di 12 settimane hanno indotto il “beiging” del tessuto adiposo bianco sottocutaneo, nonché un miglioramento della funzione delle cellule β. Mirabegron ha aumentato l’espressione proteica dei marcatori adiposi “beige” UCP1 (2,4 volte), della proteina transmembrana 26 (TMEM26) (4,2 volte) e dell’effettore A simile al DFFA che induce la morte cellulare (CIDEA) (2,4 volte) [13]. Il “beiging” del tessuto adiposo bianco sottocutaneo da parte di mirabegron può ridurre la disfunzione del tessuto adiposo, il che può migliorare la capacità ossidativa muscolare e può migliorare la funzione delle cellule β [13]. Prendendo in considerazione l’omeostasi del glucosio, il trattamento con mirabegron ha migliorato la tolleranza orale al glucosio, portando a convertire il prediabete in una normale concentrazione di glucosio, ha ridotto i livelli di emoglobina A1c e ha migliorato la sensibilità all’insulina e la funzione delle cellule β, senza influenzare la glicemia a digiuno o i livelli di insulina a digiuno e HOMA-IR. Tuttavia, i risultati delle pinze euglicemiche, che sono il gold standard per misurare la sensibilità all’insulina, hanno rivelato che il trattamento con mirabegron ha aumentato in modo coerente e significativo la velocità di infusione del glucosio di circa il 12% [13]. I livelli di lipidi plasmatici sono cambiati in modo significativo, ma, dopo il trattamento con mirabegron, è stata riscontrata una tendenza verso una riduzione del colesterolo totale [13]. Sfortunatamente, una terapia di 12 settimane non ha determinato un aumento significativo della quantità di BAT e del dispendio energetico a riposo, della perdita di peso o dei cambiamenti nella composizione corporea in tali pazienti [13].
L’effetto benefico del mirabegron, simile all’effetto dell’esposizione al freddo, sull’induzione del tessuto adiposo “beige” nel tessuto adiposo sottocutaneo umano è stato riportato anche in un altro studio condotto da Finlin et al. [14]. Hanno esposto al freddo i partecipanti alla ricerca magri e obesi o li hanno trattati con mirabegron. Il trattamento cronico con mirabegron (10 settimane; 50 mg/giorno) ha indotto UCP1 (3 volte) e TMEM26 (8,7 volte) nei soggetti obesi. Inoltre, l’espressione di UCP1 e dei marcatori degli adipociti “beige” è aumentata più che dopo 10 giorni di ripetuta esposizione al freddo [14].
Nello studio successivo, composto da 12 partecipanti obesi insulino-resistenti, Finlin et al. [9] hanno valutato la capacità del trattamento con pioglitazone (30 mg/giorno) o del trattamento con mirabegron (50 mg/giorno) in monoterapia, così come una combinazione di trattamento con pioglitazone (30 mg/giorno) e mirabegron (50 mg/giorno), di aumentare il grasso “beige” o migliorare ulteriormente il metabolismo del glucosio durante 12 settimane di terapia. Il pioglitazone è un attivatore PPARγ che può stimolare il BAT o “grasso beige”. Il trattamento con pioglitazone o la combinazione di pioglitazone e mirabegron hanno aumentato l’espressione del marcatore proteico del tessuto adiposo “beige” e migliorato la sensibilità all’insulina (misurata tramite clamp euglicemico, più efficace nella terapia combinata) e l’omeostasi del glucosio (inclusi test di tolleranza al glucosio migliorati, più efficaci nella terapia combinata), ma nessuno dei due trattamenti ha indotto il BAT o influenzato la spesa energetica nei soggetti obesi. Inoltre, non si è verificato alcun cambiamento significativo nel peso corporeo dopo il trattamento. Nonostante il fatto che mirabegron e pioglitazone somministrati separatamente abbiano indotto il “beiging” del tessuto adiposo, l’aggiunta di pioglitazone a mirabegron non ha migliorato il “beiging”, poiché il trattamento combinato ha prodotto un “beiging” inferiore rispetto a entrambi i farmaci somministrati singolarmente [9].
Sebbene i risultati preliminari degli studi sugli animali abbiano mostrato i benefici della co-somministrazione di mirabegron e metformina nella prevenzione e nel trattamento dell’obesità [2], a nostra conoscenza, l’influenza di tale terapia combinata non è stata verificata in relazione all’attività BAT, al dispendio energetico e alla perdita di peso negli esseri umani. È noto solo che non ci sono interazioni clinicamente significative tra metformina e mirabegron. Nello studio con 32 soggetti maschi sani (BMI: 18–30 kg/m2), mirabegron (160 mg somministrati una volta al giorno) non ha mostrato alcun effetto sulla farmacocinetica di metformina (500 mg somministrati due volte al giorno). La co-somministrazione di mirabegron con metformina ha determinato piccole modifiche nell’esposizione a mirabegron (AUC e Cmax diminuite del 21%). Le modifiche farmacocinetiche osservate non sono state considerate clinicamente rilevanti. Pertanto, non è necessario alcun aggiustamento del dosaggio di mirabegron quando viene co-somministrato con metformina [33].
Sebbene i dati confermino che una bassa dose di mirabegron può indurre il “beiging” del WAT sottocutaneo, è stato riportato che 50 mg di mirabegron durante il trattamento a breve termine (circa 12 settimane di terapia) non hanno alcun effetto sulla quantità di BAT, sul dispendio energetico a riposo e sulla perdita di peso. Pertanto, sono necessari studi clinici di lunga durata, con partecipanti obesi con una dose inferiore di mirabegron, per valutare se il “beiging” del tessuto adiposo si tradurrebbe in un miglioramento del dispendio energetico a riposo e in una significativa perdita di peso.
Conclusioni:
Il BAT metabolicamente attivo è stato correlato positivamente al miglioramento dell’energia, del glucosio e del metabolismo dell’intero corpo [34]. L’attivazione del BAT e l’induzione del processo di “browning” nel WAT sembrano essere un’interessante strategia terapeutica per aumentare la spesa energetica e migliorare il metabolismo. Il mirabegron, come agonista del recettore β3-adrenergico, si è rivelato efficace come attivatore del BAT, stimolatore delle cellule “beige” e regolatore dell’omeostasi metabolica sia negli studi sugli animali che negli esseri umani. Sebbene negli studi sugli animali la somministrazione di mirabegron abbia portato a un miglioramento dell’obesità, non è stata ancora dimostrata una significativa perdita di peso nei pazienti obesi dopo dosi elevate o basse del farmaco. Ciò può essere spiegato dalla durata troppo breve degli studi e dal numero esiguo di partecipanti agli studi. Inoltre, negli esseri umani, il trattamento più efficace per la stimolazione del BAT e del WAT è stato quello con dosi elevate di mirabegron; tuttavia, gli effetti collaterali cardiovascolari possono limitare l’uso di dosi superiori a quelle approvate dalla FDA per il trattamento della vescica iperattiva. Da un lato, considerando l’uso di dosi elevate di mirabegron, deve essere valutata la sicurezza a lungo termine in relazione al sistema cardiovascolare. In caso di attivazione aggravata dei recettori β1 miocardici, la somministrazione concomitante di 100-200 mg di mirabegron con un bloccante β1-AR può essere una strategia terapeutica utile per evitare effetti collaterali cardiovascolari. D’altro canto, dovrebbe essere valutato se dosi più piccole di mirabegron, ad esempio quelle approvate per la vescica iperattiva (50 mg al giorno), assunte per un periodo di tempo più lungo, saranno sufficienti a stimolare la crescita del BAT, l’imbrunimento del WAT e la termogenesi che può portare alla perdita di peso. Negli studi clinici riguardanti l’efficacia e la sicurezza del mirabegron nei pazienti con vescica iperattiva, l’influenza del mirabegron sul peso corporeo non è stata verificata. A nostra conoscenza, l’efficacia del mirabegron in relazione ai disturbi metabolici, inclusa l’obesità, nei soggetti trattati per vescica iperattiva, non è stata finora valutata.
Si potrebbe quindi ipotizzare che, il potenziale ruolo del Mirabegron nel trattamento o nella prevenzione dell’obesità dipenderebbe dai risultati della sua efficacia determinati da studi clinici a lungo termine. In caso di mancanza o insoddisfacente effetto dimagrante (rispetto ai farmaci attualmente disponibili approvati per il trattamento dell’obesità), il mirabegron potrebbe essere utilizzato per migliorare il profilo metabolico nei pazienti obesi. Se l’effetto dimagrante del mirabegron venisse confermato, il farmaco diventerebbe un’opzione alternativa agli attuali agenti anti-obesità, specialmente nei pazienti con controindicazioni o intolleranza ad altri farmaci. Inoltre, un aspetto interessante da valutare negli studi clinici sarebbe se la co-somministrazione di mirabegron e altri farmaci, come metformina, pioglitazone o altri farmaci anti-obesità attualmente utilizzati, potrebbe essere una strategia più efficace rispetto alla somministrazione di tali farmaci da soli per migliorare i profili metabolici o per trattare l’obesità. I benefici della co-somministrazione di mirabegron e metformina nella prevenzione e nel trattamento dell’obesità, dimostrati in studi sugli animali, devono essere confermati in ulteriori studi clinici. Sebbene i risultati preliminari della co-somministrazione di mirabegron e pioglitazone in partecipanti obesi non abbiano indicato alcuna influenza di tale terapia sul peso corporeo, devono essere eseguiti ulteriori studi per confermare questi risultati. Pertanto, l’introduzione di agonisti del recettore β3-adrenergico nel trattamento dell’obesità in futuro richiederà studi a lungo termine con un numero maggiore di soggetti per valutarne l’efficacia, la tollerabilità e la sicurezza.
I tessuti adiposi bruni e “beige” rimangono un bersaglio attraente per combattere le malattie metaboliche. Sono necessari ulteriori studi per confermare se la combinazione di agenti attivatori di BAT e “beige”, esercizi fisici e una dieta ipocalorica sana sarebbe una strategia di successo per ottenere la perdita di peso nei pazienti con obesità.
E per l’uso off-label nella ricomposizione corporea? I test in tal senso sono ancora scarsi e dal design spesso pessimo. Vi sono stati riscontri positivi, almeno preliminarmente parlando, con protocolli di 8-12 settimane a dosaggi di 75-100mg/die. Il dosaggio era stato settato partendo da 25mg/die per poi mantenere e osservare le risposte al dosaggio per qualche giorno [pressione, battito cardiaco ecc…]. L’uso del Mirabegron in ambito sportivo è tanto pionieristico come lo è quello dei tireomimetici. Ci vorranno ancora diversi studi per poter essere maggiormente certi di concreti vantaggi applicativi di questa molecola come PEDs.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Cypess, A.M.; Weiner, L.S.; Roberts-Toler, C.; Elia, E.F.; Kessler, S.H.; Kahn, P.A.; English, J.; Chatman, K.; Trauger, S.A.; Doria, A.; et al. Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell. Metab.2015, 21, 33–38. [Google Scholar] [CrossRef]
Zhao, X.Y.; Liu, Y.; Zhang, X.; Zhao, B.C.; Burley, G.; Yang, Z.C.; Luo, Y.; Li, A.Q.; Zhang, R.X.; Liu, Z.Y.; et al. The combined effect of metformin and mirabegron on diet-induced obesity. Med. Comm.2023, 4, e207. [Google Scholar] [CrossRef]
Nahon, K.J.; Janssen, L.G.M.; Sardjoe Mishre, A.S.D.; Bilsen, M.P.; van der Eijk, J.A.; Botani, K.; Overduin, L.A.; Ruiz, J.R.; Burakiewicz, J.; Dzyubachyk, O.; et al. The effect of mirabegron on energy expenditure and brown adipose tissue in healthy lean South Asian and Europid men. Diabetes Obes. Metab.2020, 22, 2032–2044. [Google Scholar] [CrossRef]
Hao, L.; Scott, S.; Abbasi, M.; Zu, Y.; Khan, M.S.H.; Yang, Y.; Wu, D.; Zhao, L.; Wang, S. Beneficial Metabolic Effects of Mirabegron In Vitro and in High-Fat Diet-Induced Obese Mice. J. Pharmacol. Exp. Ther.2019, 369, 419–427. [Google Scholar] [CrossRef]
Waki, H.; Yamauchi, T. Body-weight-independent glucose-lowering effect of the b3-adrenergic receptor agonist mirabegron in humans. J. Diabetes Investig.2021, 12, 689–690. [Google Scholar] [CrossRef]
Harb, E.; Kheder, O.; Poopalasingam, G.; Rashid, R.; Srinivasan, A.; Izzi-Engbeaya, C. Brown adipose tissue and regulation of human body weight. Diabetes Metab. Ress. Rev.2023, 39, e3594. [Google Scholar] [CrossRef]
Finlin, B.S.; Memetimin, H.; Zhu, B.; Confides, A.L.; Vekaria, H.J.; El Khouli, R.H.; Johnson, Z.R.; Westgate, P.M.; Chen, J.; Morris, A.J.; et al. The β3-adrenergic receptor agonist mirabegron improves glucose homeostatsis in obese humans. J. Clin. Investig.2020, 130, 2319–2331. [Google Scholar] [CrossRef]
Finlin, B.S.; Memetimin, H.; Confides, A.L.; Kasza, I.; Zhu, B.; Vekaria, H.J.; Harfmann, B.; Jones, K.A.; Johnson, Z.R.; Westgate, P.M.; et al. Human adipose beiging in response to cold and mirabegron. JCI Insight2018, 3, e121510. [Google Scholar] [CrossRef]
Dehvari, N.; Sato, M.; Bokhari, M.H.; Kalinovich, A.; Ham, S.; Gao, J.; Nguyen, H.T.M.; Whiting, L.; Mukaida, S.; Merlin, J.; et al. The metabolic effects of mirabegron are mediated primarily by β3-adrenoceptors. Pharmacol. Res. Perspect.2020, 8, e00643. [Google Scholar] [CrossRef]
Dehvari, N.; da Silva Junior, E.D.; Bengtsson, T.; Hutchinson, D.S. Mirabegron: Potential off target effects and uses beyond the bladder. Br. J. Pharmacol.2018, 175, 4072–4082. [Google Scholar] [CrossRef]
Baskin, A.S.; Linderman, J.D.; Brycha, R.J.; McGehee, S.; Anflick-Chames, E.; Cero, C.; Johnson, J.W.; O’Mara, A.E.; Fletcher, L.A.; Leitner, B.P.; et al. Regulation of Human Adipose Tissue Activation, Gallbladder Size, and Bile Acid Metabolism by a b3-Adrenergic Receptor Agonis. Diabetes2018, 67, 2113–2125. [Google Scholar] [CrossRef]
Pereas Valgas da Silva, C.; Calmasini, F.; Alexandre, E.C.; Raposo, H.F.; Delbin, M.A.; Monica, F.Z.; Zanesco, A. The effects of mirabegron on obesity-induced inflammation and insulin resistance are associated with brown adipose tissue activation but not beiging in the subcutaneous white adipose tissue. Clin. Exp. Pharmacol. Physiol.2021, 48, 1477–1487. [Google Scholar] [CrossRef]
Arch, J.R.S. Challenges in β3-adrenoceptor agonist drug development. Ther. Adv. Endocrinol. Metab.2011, 2, 59–64. [Google Scholar] [CrossRef]
Liu, X.; Pérusse, F.; Bukowiecki, L.J. Mechanisms of the antidiabetic effects of the beta 3-adrenergic agonist CL-316243 in obese Zucker-ZDF rats. Am. J. Physiol.1998, 274, R1212–R1219. [Google Scholar]
Takasu, T.; Ukai, M.; Sato, S.; Matsui, T.; Nagase, I.; Maruyama, T.; Sasamata, M.; Miyata, K.; Uchida, H.; Yamaguchi, O. Effect of (R)-2-(2-aminothiazol-4-yl)-4′-{2-[(2-hydroxy-2-phenylethyl)amino]ethyl} acetanilide (YM178), a novel selective beta3-adrenoceptor agonist, on bladder function. J. Pharmacol. Exp. Ther.2007, 321, 642–647. [Google Scholar] [CrossRef]
Igawa, Y.; Michel, M.C. Pharmacological profile of β3-adrenoceptor agonists in clinical development for the treatment of overactive bladder syndrome. Naunyn. Schmiedebergs. Arch. Pharmacol.2013, 386, 177–183. [Google Scholar] [CrossRef] [PubMed]
Krhut, J.; Martan, A.; Zachoval, R.; Hanus, T.; Svabik, K.; Zvara, P. Impact of body mass index on treatment efficacy of mirabegron for overactive bladder in females. Eur. J. Obstet. Gynecol. Reprod. Biol.2016, 196, 64–68. [Google Scholar] [CrossRef] [PubMed]
Hatanaka, T.; Ukai, M.; Watanabe, M.; Someya, A.; Ohtake, A.; Suzuki, M.; Ueshima, K.; Sato, S.; Sasamata, M. In vitro and in vivo pharmacological profile of the selective β3-adrenoceptor agonist mirabegron in rats. Naunyn. Schmiedebergs. Arch. Pharmacol.2013, 386, 247–253. [Google Scholar] [CrossRef]
Nagiri, C.; Kobayashi, K.; Tomita, A.; Kato, M.; Kobayashi, K.; Yamashita, K.; Nishizawa, T.; Inoue, A.; Shihoya, W.; Nureki, O. Cryo-EM structure of the β3-adrenergic receptor reveals the molecular basis of subtype selectivity. Mol. Cell.2021, 81, 3205–3215. [Google Scholar] [CrossRef] [PubMed]
Brucker, B.M.; King, J.; Mudd, P.N., Jr.; McHale, K. Selectivity and Maximum Response of Vibegron and Mirabegron for β3-Adrenergic Receptors. Curr. Ther. Res. Clin. Exp.2022, 96, 100674. [Google Scholar] [CrossRef]
Alexandre, E.C.; Kiguti, L.R.; Calmasini, F.B.; Silva, F.H.; da Silva, K.P.; Ferreira, R.; Ribeiro, C.A.; Mónica, F.Z.; Pupo, A.S.; Antunes, E. Mirabegron relaxes urethral smooth muscle by a dual mechanism involving β3 -adrenoceptor activation and α1 -adrenoceptor blockade. Br. J. Pharmacol.2016, 173, 415–428. [Google Scholar] [CrossRef]
De Stefano, A.; Schinzari, F.; Di Daniele, N.; Sica, G.; Gentileschi, P.; Vizioli, G.; Cardillo, C.; Tesauro, M. Mirabegron relaxes arteries from human visceral adipose tissue through antagonism of α1-adrenergic receptors. Vascul. Pharmacol.2022, 146, 107094. [Google Scholar] [CrossRef]
Lowell, B.B.; S-Susulic, V.; Hamann, A.; Lawitts, J.A.; Himms-Hagen, J.; Boyer, B.B.; Kozak, L.P.; Flier, J.S. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature1993, 366, 740–742. [Google Scholar] [CrossRef]
Hamann, A.; Flier, J.S.; Lowell, B.B. Decreased brown fat markedly enhances susceptibility to diet-induced obesity, diabetes, and hyperlipidemia. Endocrinology1996, 137, 21–29. [Google Scholar] [CrossRef] [PubMed]
Lv, Z.; Guo, Y. Metformin and its benefits for various diseases. Front. Endocrinol.2020, 11, 191. [Google Scholar] [CrossRef] [PubMed]
Groen-Wijnberg, M.; van Dijk, J.; Krauwinkel, W.; Kerbusch, V.; Meijer, J.; Tretter, R.; Zhang, W.; van Gelderen, M. Pharmacokinetic Interactions Between Mirabegron and Metformin, Warfarin, Digoxin or Combined Oral Contraceptives. Eur. J. Drug Metab. Pharmacokinet.2017, 42, 417–429. [Google Scholar] [CrossRef] [PubMed]
Bel, J.C.; Tai, T.C.; Khaper, N.; Lees, S.J. Mirabegron: The most promising adipose tissue beiging agent. Physiol. Rep.2021, 9, e14779. [Google Scholar] [CrossRef]
I Glucocorticoidi (o, meno comunemente, glucocorticosteroidi) sono una classe di corticosteroidi, ovvero una classe di ormoni steroidei. I Glucocorticoidi sono corticosteroidi che si legano al recettore dei glucocorticoidi[1], presente in quasi tutte le cellule animali vertebrate. Il nome “Glucocorticoide” è un portmanteau (glucosio + corteccia + steroide) ed è composto dal suo ruolo nella regolazione del metabolismo del glucosio, dalla sintesi nella corteccia surrenale e dalla sua struttura steroidea.
I Glucocorticoidi fanno parte del meccanismo di feedback del sistema immunitario, che riduce alcuni aspetti della funzione immunitaria, come l’infiammazione. Sono quindi utilizzati in medicina per trattare le malattie causate da un sistema immunitario iperattivo, come allergie, asma, malattie autoimmuni e sepsi. I Glucocorticoidi hanno molti effetti diversi, come la pleiotropia, tra cui effetti collaterali potenzialmente dannosi.[2] Inoltre, interferiscono con alcuni meccanismi anomali delle cellule tumorali, per cui vengono utilizzati in dosi elevate per il trattamento del cancro. Ciò include effetti inibitori sulla proliferazione dei linfociti, come nel trattamento di linfomi e leucemie, e l’attenuazione degli effetti collaterali dei farmaci antitumorali.
I glucocorticoidi agiscono sulle cellule legandosi al Recettore dei Glucocorticoidi (GR). Il complesso recettore glucocorticoide-glucocorticoide attivato regola l’espressione di proteine antinfiammatorie nel nucleo (processo noto come transattivazione) e reprime l’espressione di proteine pro-infiammatorie nel citosol impedendo la traslocazione di altri fattori di trascrizione dal citosol al nucleo (transrepressione).[2]
Il Recettore dei Glucocorticoidi (GR o GCR), noto anche come NR3C1 (sottofamiglia 3 del recettore nucleare, gruppo C, membro 1), è il recettore a cui si legano il Cortisolo e altri Glucocorticoidi.
I glucocorticoidi si distinguono dai mineralocorticoidi e dagli steroidi sessuali per i loro specifici recettori, cellule bersaglio ed effetti. In termini tecnici, il termine “corticosteroide” si riferisce sia ai glucocorticoidi che ai mineralocorticoidi (poiché entrambi sono mimici degli ormoni prodotti dalla corteccia surrenale), ma è spesso usato come sinonimo di “glucocorticoide”. I glucocorticoidi sono prodotti principalmente nella zona fascicolata della corteccia surrenale, mentre i mineralocorticoidi sono sintetizzati nella zona glomerulosa.
Il Cortisolo (o idrocortisone) è il più importante glucocorticoide umano. È essenziale per la vita e regola o supporta una serie di importanti funzioni cardiovascolari, metaboliche, immunologiche e omeostatiche. Gli aumenti delle concentrazioni di glucocorticoidi sono parte integrante della risposta allo stress e sono i biomarcatori più comunemente utilizzati per misurare lo stress.[3] I glucocorticoidi hanno anche numerose funzioni non correlate allo stress e le concentrazioni di glucocorticoidi possono aumentare in risposta al piacere o all’eccitazione.[4] Sono disponibili diversi glucocorticoidi sintetici, ampiamente utilizzati nella pratica medica generale e in numerose specialità, come terapia sostitutiva in caso di carenza di glucocorticoidi o per sopprimere il sistema immunitario dell’organismo.
Si sospetta che gli atleti utilizzino i Glucocorticoidi per migliorare le prestazioni atletiche fin dagli anni ’60 (5). I glucocorticoidi possono migliorare le prestazioni fisiche attraverso diverse vie, tra cui una maggiore disponibilità di substrati metabolici attraverso un aumento della lipolisi (6), della proteolisi (7) e della disponibilità di glucosio (5). I Glucocorticoidi possono anche avere effetti immunosoppressivi e antinfiammatori che possono impedire al sistema immunitario di reagire in modo eccessivo a seguito di danni muscolari indotti dall’esercizio fisico (8), e il Cortisolo sembra preparare l’organismo per l’esercizio successivo (9). Inoltre, i glucocorticoidi possono stimolare i recettori cerebrali dei glucocorticoidi, determinando una riduzione del dolore muscolare durante l’esercizio, un aumento della soglia della fatica e risposte edoniche positive, che possono tradursi in un aumento delle prestazioni fisiche (10). I glucocorticoidi hanno un elevato potenziale di effetti avversi, tra cui il deperimento muscolare, e questi effetti dipendono da molteplici fattori come il tipo di glucocorticoide, la durata del trattamento, la dose e la via di somministrazione (11). Pertanto, gli atleti che assumono glucocorticoidi per migliorare le prestazioni probabilmente preferiscono periodi di somministrazione più brevi.
L’Associazione Mondiale Antidoping (WADA) aggiorna annualmente la sua lista proibita, un elenco di sostanze e metodi vietati negli sport d’élite. La lista proibita, in vigore dal 1° gennaio 2022, vieta tutti i glucocorticoidi nelle competizioni quando sono somministrati per via orale, rettale o iniettabile, poiché queste forme di somministrazione sono considerate ad effetto sistemico. Altre vie di somministrazione, come l’applicazione topica per inalazione e le iniezioni locali, sono approvate nelle competizioni, poiché si ritiene che abbiano un minore potenziale di miglioramento delle prestazioni. L’uso dei glucocorticoidi al di fuori delle competizioni è approvato (12). Per aiutare i medici a curare gli atleti e a rispettare le norme antidoping, dal 2022 la WADA raccomanda specifici periodi di wash out (tempo dall’ultima dose al giorno prima della competizione) per i diversi tipi di glucocorticoidi e le diverse vie di somministrazione. Dal 2022 la WADA ha anche introdotto livelli di segnalazione urinaria specifici per i diversi tipi di glucocorticoidi, in quanto i diversi glucocorticoidi hanno un’ampia variazione nel tempo di eliminazione (13).
Dagli anni 90 si è studiato l’effetto dei glucocorticoidi sulla frequenza cardiaca e sul consumo di ossigeno durante intervalli di corsa ad alta intensità. Da allora diversi studi RCT hanno analizzato l’effetto dei glucocorticoidi sulla prestazione fisica submassimale e massimale. Poiché le modalità di somministrazione, il tipo di glucocorticoide, la durata del trattamento, la dose, la popolazione e il protocollo di esercizio possono variare tra gli studi, essi giungono a conclusioni diverse per quanto riguarda l’effetto dei glucocorticoidi sulla prestazione fisica, tuttavia sono soprattutto gli studi che indagano l’effetto di alte dosi orali somministrate nel tempo sulla resistenza in bicicletta fino all’esaurimento a mostrare effetti. L’effetto dei glucocorticoidi sulla prestazione fisica e sul metabolismo dei soggetti sani è stato esaminato in precedenza (5, 10), ma senza una ricerca sistematica della letteratura e senza combinare i risultati degli studi inclusi nell’analisi statistica. Pertanto, in tale sede, cercherò anche di fare chiarezza sull’effetto dei glucocorticoidi sulla prestazione massimale o submassimale in soggetti sani.
Steroidogenesi dei Glucocorticoidi:
La steroidogenesi è il processo biologico attraverso il quale gli steroidi vengono generati dal colesterolo e trasformati in altri steroidi.[14] Le vie della steroidogenesi differiscono tra le specie. Le principali classi di ormoni steroidei, con i loro membri e le loro funzioni principali, sono i progestinici, i corticosteroidi (corticoidi), gli androgeni e gli estrogeni.[15][16] La steroidogenesi umana dei corticosteroidi avviene nella Corteccia Surrenale.
Steroidogenesi che mostra i Glucocorticoidi nell’ellisse verde a destra con l’esempio primario del Cortisolo. Non è un gruppo strettamente delimitato, ma un continuum di strutture con effetto glucocorticoide crescente.
La Corteccia Surrenale è la regione più esterna e anche la parte più grande della ghiandola surrenale. È divisa in tre zone distinte: zona glomerulosa, zona fascicolata e zona reticolare. Ogni zona è responsabile della produzione di ormoni specifici. È anche un sito secondario di sintesi degli androgeni.[17]
Corteccia Surrenale
Zona Glomerulosa
Colorazione H&E della Corteccia Surrenale. La Zona Glomerulare è lo strato più esterno, sotto la capsula renale (vicino all’indicatore)
Lo strato più esterno, la zona glomerulosa, è il sito principale per la produzione di aldosterone, un mineralcorticoide. La sintesi e la secrezione di aldosterone sono regolate principalmente dal sistema renina-angiotensina-aldosterone. Le cellule della zona glomerulosa esprimono un enzima specifico, l’aldosterone sintasi (noto anche come CYP11B2).[18][19] L’aldosterone è ampiamente responsabile della regolazione a lungo termine della pressione sanguigna. [Gli effetti dell’aldosterone si manifestano nel tubulo contorto distale e nel dotto collettore del rene, dove provoca un aumento del riassorbimento del sodio e una maggiore escrezione di potassio (da parte delle cellule principali) e di ioni idrogeno (da parte delle cellule intercalate del dotto collettore).[20] La ritenzione di sodio è anche una risposta del colon distale e delle ghiandole sudoripare alla stimolazione dei recettori dell’aldosterone. Sebbene la produzione sostenuta di aldosterone richieda un ingresso persistente di calcio attraverso canali del Ca2+ attivati a basso voltaggio, le cellule isolate della zona glomerulosa sono considerate non eccitabili, con tensioni di membrana registrate troppo iperpolarizzate per consentire l’ingresso di canali del Ca2+.[21]
La secrezione di aldosterone è stimolata anche dall’ormone adrenocorticotropo (ACTH).[22]
Le cellule della zona glomerulosa non esprimono l’11β-idrossilasi e la 17α-idrossilasi. Per questo motivo la zona glomerulosa non può sintetizzare cortisolo, corticosterone o ormoni sessuali (androgeni). [23] L’espressione di proteine specifiche per i neuroni nelle cellule della zona glomerulosa dei tessuti adrenocorticali umani è stata prevista e riportata da diversi autori [24][25][26] ed è stato suggerito che l’espressione di proteine come la molecola di adesione delle cellule neuronali (NCAM) nelle cellule della zona glomerulosa rifletta la caratteristica rigenerativa di queste cellule, che perderebbero l’immunoreattività della NCAM dopo essersi spostate nella zona fascicolata. [24][27] Tuttavia, insieme ad altri dati sulle proprietà neuroendocrine delle cellule della zona glomerulosa, l’espressione di NCAM potrebbe riflettere una differenziazione neuroendocrina di queste cellule.[24]
Zona Fasciculata
Zona Fasciculata
Situate tra la glomerulosa e la reticolare, le cellule della zona fascicolata sintetizzano e secernono glucocorticoidi (come l’11-deossicorticosterone, il corticosterone e il cortisolo), oltre a piccole quantità di androgeni ed estrogeni surrenalici.[28] La zona fascicolata ha una maggiore attività di 3β-idrossisteroide deidrogenasi rispetto alla zona reticolare. Pertanto, la zona fascicolata produce più 11-deossicorticosterone, corticosterone e cortisolo.[23] Il principale ormone che stimola la secrezione di cortisolo nell’uomo è l’ACTH, rilasciato dall’ipofisi anteriore.[22] È stato dimostrato che la capacità steroidogenica della zona fascicolata aumenta durante la malattia nei neonati.[22]
Zona Reticolare
Zona Reticolare
La zona reticolare, lo strato corticale più interno, produce gli androgeni surrenalici, oltre a piccole quantità di estrogeni e alcuni glucocorticoidi.[28] La zona reticolare possiede una quantità maggiore di cofattori necessari per l’attività della 17,20-liasi della 17α-idrossilasi rispetto alla zona fascicolata. Pertanto, la zona reticolare produce più androgeni,[23] soprattutto deidroepiandrosterone (DHEA), DHEA solfato (DHEA-S) e androstenedione (il precursore del Testosterone e DHT) nell’uomo. La secrezione di DHEAS è stimolata anche dall’ACTH.[22]
Come abbiamo visto, I glucocorticoidi sono prodotti principalmente nella Zona Fascicolata.[23]
Il precursore degli steroidi sintetizzati nella corteccia surrenale è il colesterolo che viene immagazzinato nelle vescicole. Il colesterolo può essere sintetizzato de novo nella corteccia surrenale. Tuttavia, la fonte principale di colesterolo sembra essere il colesterolo assunto dalle lipoproteine circolanti. [29]
I passaggi fino a questo punto avvengono in molti tessuti produttori di steroidi. Le fasi successive per generare aldosterone e cortisolo, tuttavia, avvengono principalmente nella corteccia surrenale:
Progesterone → (idrossilazione a C21) → 11-Deossicorticosterone → (due ulteriori idrossilazioni a C11 e C18) → Aldosterone
Progesterone → (idrossilazione a C17) → 17-alfa-idrossiprogesterone → (idrossilazione a C21) → 11-Deossicortisolo → (idrossilazione a C11) → Cortisolo
Fasi della sintesi dell’ormone steroideo surrenale
Effetto sistemico dei Glucocorticoidi:
Gli effetti dei glucocorticoidi possono essere ampiamente classificati in due categorie principali:
immunologici
metabolici.
Inoltre, i glucocorticoidi svolgono ruoli importanti nello sviluppo fetale e nell’omeostasi dei fluidi corporei.
Immunità
Come già accennato, i glucocorticoidi funzionano anche attraverso l’interazione con il recettore dei glucocorticoidi:
Aumentano l’espressione di proteine antinfiammatorie.
Riducono l’espressione di proteine pro-infiammatorie.
Micrografia elettronica a scansione di un globulo rosso (sinistra), una piastrina (centro) e un linfocita T (destra); colorato
È stato dimostrato che i glucocorticoidi svolgono un ruolo nello sviluppo e nell’omeostasi dei linfociti T. Questo è stato dimostrato in transgenici. Ciò è stato dimostrato in topi transgenici con una maggiore o minore sensibilità della linea delle cellule T ai glucocorticoidi.[30]
Metabolismo
Nello stato di digiuno, il cortisolo stimola diversi processi che servono collettivamente ad aumentare e mantenere le normali concentrazioni di glucosio nel sangue.
Effetti metabolici:
Stimolazione della gluconeogenesi, in particolare nel fegato: Questa via porta alla sintesi del glucosio a partire da substrati non esosi, come gli aminoacidi e il glicerolo proveniente dalla scissione dei trigliceridi, ed è particolarmente importante nei carnivori e in alcuni erbivori. L’aumento dell’espressione degli enzimi coinvolti nella gluconeogenesi è probabilmente la funzione metabolica più nota dei glucocorticoidi.
Mobilitazione di aminoacidi dai tessuti extraepatici: Questi servono come substrati per la gluconeogenesi.
Inibizione della captazione del glucosio nel tessuto muscolare e adiposo: Un meccanismo per conservare il glucosio
Stimolazione della demolizione dei grassi nel tessuto adiposo: Gli acidi grassi rilasciati dalla lipolisi vengono utilizzati per la produzione di energia in tessuti come il muscolo e il glicerolo rilasciato fornisce un altro substrato per la gluconeogenesi.
L’aumento della ritenzione di sodio e dell’escrezione di potassio porta a ipernatremia e ipokaliemia[31].
Aumento della concentrazione di emoglobina, probabilmente dovuto all’ostacolo dell’ingestione di globuli rossi da parte di macrofagi o altri fagociti[32].
Aumento dell’acido urico urinario[33]
Aumento del calcio urinario e ipocalcemia[34]
Alcalosi[35]
Leucocitosi[36]
Il Cortisolo innesca una cascata di eventi che influenzano l’omeostasi del glucosio. Il fegato, i muscoli scheletrici, i tessuti adiposi bianchi e il pancreas svolgono un ruolo chiave nell’assicurare un apporto continuo di energia utilizzabile per la risposta di lotta/fuga.
Livelli eccessivi di glucocorticoidi derivanti dalla somministrazione di farmaci o dall’iperadrenocorticismo hanno effetti su molti sistemi. Alcuni esempi includono l’inibizione della formazione ossea, la soppressione dell’assorbimento del calcio (entrambi possono portare all’osteoporosi), il ritardo nella guarigione delle ferite, la debolezza muscolare e l’aumento del rischio di infezioni. Queste osservazioni suggeriscono una moltitudine di ruoli fisiologici meno drammatici per i glucocorticoidi.[30]
Eccitazione e sfera cognitiva
Principali scissure e lobi del cervello visti lateralmente (il lobo frontale è mostrato in blu).
I glucocorticoidi agiscono sull’ippocampo, sull’amigdala e sui lobi frontali. Insieme all’adrenalina, favoriscono la formazione di ricordi flashbulb di eventi associati a forti emozioni, sia positive che negative.[36] Ciò è stato confermato da studi in cui il blocco dell’attività dei glucocorticoidi o della noradrenalina ha compromesso il richiamo di informazioni emotivamente rilevanti. Ulteriori fonti hanno dimostrato che i soggetti il cui apprendimento della paura è stato accompagnato da alti livelli di cortisolo hanno avuto un migliore consolidamento di questa memoria (questo effetto è stato più importante negli uomini). L’effetto che i glucocorticoidi hanno sulla memoria può essere dovuto a un danno specifico all’area CA1 della formazione dell’ippocampo.
In diversi studi sugli animali, lo stress prolungato (che causa aumenti prolungati dei livelli di glucocorticoidi) ha mostrato la distruzione dei neuroni nell’area dell’ippocampo del cervello, che è stata collegata a prestazioni di memoria inferiori.[32][37][33]
Una rappresentazione grafica della curva di Yerkes-Dodson
È stato inoltre dimostrato che i glucocorticoidi hanno un impatto significativo sulla vigilanza (disturbo da deficit di attenzione) e sulla cognizione (memoria). Questo sembra seguire la curva di Yerkes-Dodson, in quanto gli studi hanno dimostrato che i livelli circolanti di glucocorticoidi rispetto alle prestazioni della memoria seguono un andamento a U rovesciata, proprio come la curva di Yerkes-Dodson. Ad esempio, il potenziamento a lungo termine (LTP, il processo di formazione dei ricordi a lungo termine) è ottimale quando i livelli di glucocorticoidi sono leggermente elevati, mentre si osserva una significativa riduzione dell’LTP dopo la surrenalectomia (stato di basso livello di glucocorticoidi) o dopo la somministrazione di glucocorticoidi esogeni (stato di alto livello di glucocorticoidi). Livelli elevati di glucocorticoidi migliorano la memoria per gli eventi emotivamente eccitanti, ma portano più spesso a una scarsa memoria per il materiale non correlato alla fonte di stress/eccitazione emotiva.[38] In contrasto con gli effetti di potenziamento dose-dipendenti dei glucocorticoidi sul consolidamento della memoria, è stato dimostrato che questi ormoni dello stress inibiscono il recupero di informazioni già memorizzate. [È stato dimostrato che l’esposizione a lungo termine a farmaci glucocorticoidi, come quelli contro l’asma e gli antinfiammatori, crea deficit di memoria e attenzione sia durante che, in misura minore, dopo il trattamento,[39][40] una condizione nota come “demenza da steroidi”.[41]
Omeostasi dei fluidi corporei
I glucocorticoidi potrebbero agire a livello centrale e periferico per contribuire alla normalizzazione del volume dei liquidi extracellulari regolando l’azione dell’organismo nei confronti del peptide natriuretico atriale (ANP). A livello centrale, i glucocorticoidi potrebbero inibire l’assunzione di acqua indotta dalla disidratazione;[42] a livello periferico, i glucocorticoidi potrebbero indurre una potente diuresi.[43]
Metabolismo dei Glucocorticoidi. La secrezione di Glucocorticoidi da parte della ghiandola surrenale è regolata dall’asse HPA tramite secrezione di ACTH. Il cortisolo plasmatico principale (F) è legato alle proteine con una frazione libera del 4-5%. Il Cortisone plasmatico (E) è nella forma libera non legata. L’equilibrio di cortisolo e cortisone tra plasma e tessuti è illustrato con le frecce bidirezionali tratteggiate. È anche raffigurato il metabolismo dei GC tessuto-specifici. I GC sono metabolizzati principalmente nel fegato e i metaboliti sono escreti nelle urine. Sono mostrati solo i tessuti rilevanti per la sindrome metabolica. THE, tetraidrocortisone; THF, tetraidrocortisolo.
Meccanismi d’azione dei Glucocorticoidi:
Transattivazione
I glucocorticoidi si legano al recettore citosolico dei glucocorticoidi, un tipo di recettore nucleare che viene attivato dal legame con il ligando. Dopo che un ormone si lega al recettore corrispondente, il complesso appena formato si trasloca nel nucleo della cellula, dove si lega agli elementi di risposta ai glucocorticoidi nella regione promotrice dei geni bersaglio, determinando la regolazione dell’espressione genica. Questo processo viene comunemente definito attivazione trascrizionale o transattivazione.[44][45]
Le proteine codificate da questi geni regolati hanno un’ampia gamma di effetti, tra cui, ad esempio:[45]
L’Annessina [Lipocortina I]
Antinfiammatori – lipocortina I, proteina legante p11/calpactina, inibitore secretorio della proteasi leucocitaria 1 (SLPI) e fosfatasi della proteina chinasi attivata dal mitogeno (MAPK fosfatasi).
Aumento della gluconeogenesi – glucosio 6-fosfatasi e tirosina aminotransferasi
Transrepressione
Il meccanismo opposto è chiamato repressione trascrizionale o transrepressione. Secondo la concezione classica di questo meccanismo, il recettore dei glucocorticoidi attivato si lega al DNA nello stesso sito in cui si legherebbe un altro fattore di trascrizione, impedendo la trascrizione di geni che vengono trascritti tramite l’attività di quel fattore.[44][45] Sebbene ciò avvenga, i risultati non sono coerenti per tutti i tipi di cellule e per tutte le condizioni; non esiste un meccanismo generale e generalmente accettato per la transrepressione.[45]
Meccanismo d’azione di NF-κB.
Si stanno scoprendo nuovi meccanismi in cui la trascrizione viene repressa, ma il recettore dei glucocorticoidi attivato non interagisce con il DNA, bensì direttamente con un altro fattore di trascrizione, interferendo con esso, o con altre proteine che interferiscono con la funzione di altri fattori di trascrizione. Quest’ultimo meccanismo sembra essere il modo più probabile in cui il recettore glucocorticoide attivato interferisce con NF-κB, ossia reclutando istone deacetilasi, che deacetilano il DNA nella regione del promotore portando alla chiusura della struttura cromatinica in cui NF-κB deve legarsi.[44][45]
Attività non-genomica
Il recettore glucocorticoide attivato ha effetti che, come è stato dimostrato sperimentalmente, sono indipendenti da qualsiasi effetto sulla trascrizione e possono essere dovuti solo al legame diretto del recettore glucocorticoide attivato con altre proteine o con l’mRNA.[44][45]
Effetti genomici e non genomici dei Glucocorticoidi. Trans-attivazione: l’effetto genomico del GC dopo il legame del GR al suo elemento di risposta positiva causa una maggiore trascrizione di proteine antinfiammatorie, ad esempio, lipocortina-1, IL-10, IL-12, MAPK fosfatasi I e IκB. Trans-repressione: l’interazione molecola-molecola tra GR attivato e fattori di trascrizione pro-infiammatori, ad esempio, AP-1 o NF-κB causa una riduzione della trascrizione di mediatori pro-infiammatori, ad esempio, Il-2, IL-3, IL-4, IL-5, IL-6, IL-13, IL-15, TNF-α e VCAM-a.
Ad esempio, la chinasi Src, che si lega al recettore glucocorticoide inattivo, viene rilasciata quando un glucocorticoide si lega al recettore glucocorticoide e fosforila una proteina che a sua volta sposta una proteina adattatrice da un recettore importante nell’infiammazione, il fattore di crescita epidermico, riducendone l’attività, che a sua volta si traduce in una riduzione della creazione di acido arachidonico, una molecola proinfiammatoria chiave. Questo è uno dei meccanismi con cui i glucocorticoidi hanno un effetto antinfiammatorio.[44]
Farmacologia dei Glucocorticoidi:
Fludrocortisone Acetato
Per uso terapeutico sono stati creati diversi Glucocorticoidi sintetici, alcuni molto più potenti del Cortisolo. Si differenziano sia per la farmacocinetica (fattore di assorbimento, emivita, volume di distribuzione, clearance) che per la farmacodinamica (ad esempio la capacità di attività mineralcorticoide: ritenzione di sodio (Na+) e acqua; fisiologia renale). Poiché permeano facilmente l’intestino, vengono somministrati principalmente per os (per bocca), ma anche con altri metodi, ad esempio per via topica sulla pelle. Oltre il 90% di essi lega diverse proteine plasmatiche, anche se con una diversa specificità di legame. I glucocorticoidi endogeni e alcuni corticoidi sintetici hanno un’elevata affinità con la proteina Transcortina (detta anche globulina legante i corticosteroidi), mentre tutti legano l’albumina. Nel fegato, vengono rapidamente metabolizzati mediante coniugazione con un solfato o un acido glucuronico e vengono secreti nelle urine.
La potenza dei Glucocorticoidi, la durata dell’effetto e la sovrapposizione della potenza dei mineralocorticoidi variano. Il cortisolo è lo standard di confronto per la potenza dei glucocorticoidi. Idrocortisone è il nome utilizzato per le preparazioni farmaceutiche di cortisolo.
I dati riportati di seguito si riferiscono alla somministrazione orale. La potenza orale può essere inferiore a quella parenterale perché quantità significative (fino al 50% in alcuni casi) possono non raggiungere la circolazione. Il Fludrocortisone Acetato e il desossicorticosterone acetato sono, per definizione, mineralocorticoidi piuttosto che glucocorticoidi, ma hanno una potenza glucocorticoide minore e sono inclusi in questa tabella per fornire una prospettiva sulla potenza dei mineralocorticoidi.
Usi terapeutici:
I Glucocorticoidi possono essere utilizzati a basse dosi nell’insufficienza surrenalica. A dosi molto più elevate, i glucocorticoidi per via orale o inalatoria sono utilizzati per sopprimere vari disturbi allergici, infiammatori e autoimmuni. I glucocorticoidi per via inalatoria sono il trattamento di seconda linea per l’asma. Sono anche somministrati come immunosoppressori post-trapianto per prevenire il rigetto acuto del trapianto e la malattia del trapianto contro l’ospite. Tuttavia, non prevengono un’infezione e inibiscono anche i successivi processi riparativi. Nuove evidenze hanno dimostrato che i glucocorticoidi potrebbero essere utilizzati nel trattamento dell’insufficienza cardiaca per aumentare la responsività renale ai diuretici e ai peptidi natriuretici. I glucocorticoidi sono storicamente utilizzati per alleviare il dolore nelle condizioni infiammatorie.[46][47][48] Tuttavia, i corticosteroidi mostrano un’efficacia limitata nell’alleviare il dolore e potenziali eventi avversi per il loro uso nelle tendinopatie.[49]
Terapia Sostitutiva
Cortisolo
Qualsiasi glucocorticoide può essere somministrato in una dose che fornisce all’incirca gli stessi effetti glucocorticoidi della normale produzione di cortisolo; si parla di dosaggio fisiologico, sostitutivo o di mantenimento. Si tratta di circa 6-12mg/m2/die di Idrocortisone (m2 si riferisce all’area di superficie corporea (BSA), ed è una misura delle dimensioni del corpo; la BSA di un uomo medio è di 1,9 m2).
Gli usi clinici dei glucocorticoidi comprendono quindi:
Terapia Immunosoppressiva: I glucocorticoidi causano immunosoppressione e la componente terapeutica di questo effetto è principalmente la diminuzione della funzione e del numero di linfociti, compresi i linfociti B e i linfociti T.
Terapia Anti-Infiammatoria: I glucocorticoidi sono potenti antinfiammatori, indipendentemente dalla causa dell’infiammazione; il loro meccanismo antinfiammatorio primario è la sintesi della lipocortina-1 (annexin-1). La lipocortina-1 sopprime la fosfolipasi A2, bloccando così la produzione di eicosanoidi, e inibisce vari eventi infiammatori dei leucociti (adesione epiteliale, emigrazione, chemiotassi, fagocitosi, esplosione respiratoria, ecc.) In altre parole, i glucocorticoidi non solo sopprimono la risposta immunitaria, ma inibiscono anche i due principali prodotti dell’infiammazione, le prostaglandine e i leucotrieni. Inibiscono la sintesi delle prostaglandine a livello della fosfolipasi A2 e a livello della cicloossigenasi/PGE isomerasi (COX-1 e COX-2),[50] quest’ultimo effetto è molto simile a quello dei FANS, potenziando così l’effetto antinfiammatorio. Inoltre, i glucocorticoidi sopprimono anche l’espressione della ciclossigenasi.[51]
Trattamento del Iperaldosteronismo: I glucocorticoidi possono essere utilizzati nella gestione dell’iperaldosteronismo familiare di tipo 1. Non sono efficaci, tuttavia, per l’uso nella condizione di tipo 2.
Trattamento insufficienza cardiaca: I glucocorticoidi possono essere utilizzati nel trattamento dell’insufficienza cardiaca scompensata per potenziare la reattività renale ai diuretici, in particolare nei pazienti con insufficienza cardiaca con resistenza diuretica refrattaria a dosi elevate di diuretici dell’ansa.[52][53][54][55][56][57][58]
Meccanismi di resistenza ai corticosteroidi
La resistenza agli usi terapeutici dei glucocorticoidi può verificarsi in un certo numero di pazienti e presentare delle difficoltà; ad esempio, il 25% dei casi di asma grave può non rispondere agli steroidi. Questo può essere il risultato di una predisposizione genetica, dell’esposizione continua alla causa dell’infiammazione (come gli allergeni), di fenomeni immunologici che bypassano i glucocorticoidi, di disturbi farmacocinetici (assorbimento incompleto o escrezione o metabolismo accelerati) e di infezioni respiratorie virali e/o batteriche.[59][60]
Glucocorticoidi e Sport:
Come abbiamo visto, i Glucocorticoidi sono una delle classi di farmaci più ampiamente utilizzate ed efficaci nella popolazione generale e sono disponibili in una varietà di formulazioni farmaceutiche (ad esempio, iniezioni, compresse, creme, colliri, gocce auricolari, inalatori e spray nasali). Somministrati sia per i loro effetti sistemici che locali, i Glucocorticoidi sono utilizzati a livello globale in una vasta gamma di specialità cliniche, principalmente per le loro proprietà antinfiammatorie e immunosoppressive. In alcuni contesti, l’uso medico dei Glucocorticoidi orali sembra essere aumentato negli ultimi anni poiché questi sono un’alternativa accessibile e conveniente ai farmaci mirati ma più costosi. La prevalenza dell’uso sistemico prevalentemente per uso a breve termine varia tra l’1% e il 3%, sebbene abbia raggiunto il 17,1% in un recente studio sugli adulti in Francia.(https://bjsm.bmj.com) Nelle popolazioni di atleti, vi è una maggiore prevalenza di lesioni muscoloscheletriche e asma, e pertanto un frequente uso legittimo di Glucocorticoidi terapeutici non sarebbe sorprendente. Tuttavia, vi è una scarsità di stime di prevalenza nelle popolazioni di atleti. Un’analisi di TUE abbreviate in cui il CIO è stato informato dell’uso di Glucocorticoidi da parte degli atleti prima dei Giochi olimpici negli anni ’90 e nei primi anni 2000 suggerisce che almeno il 5% al 12% degli atleti d’élite competitivi è stato trattato con Glucocorticoidi tramite tutte le vie, prevalentemente inalatoria. In un recente sondaggio internazionale non pubblicato di medici che lavorano con atleti d’élite, oltre l’85% ha riferito di aver somministrato almeno occasionalmente Glucocorticoidi iniettabili come parte della loro normale pratica (comunicazione personale, Dr David Hughes, Australian Institute of Sport).
I Glucocorticoidi, somministrati tramite determinate vie, sono stati proibiti per la prima volta nello sport dal CIO nel 1985 e sono stati proibiti dalla WADA sin dalla sua Lista iniziale, pubblicata nel 2004. Le sostanze o i metodi sono considerati per l’inclusione nella Lista se soddisfano due dei tre criteri seguenti come stabilito dal Codice mondiale antidoping:
potenziale di migliorare o migliorano le prestazioni sportive;
rappresentano un rischio effettivo o potenziale per la salute dell’atleta;
violano lo spirito dello sport. I Glucocorticoidi sono proibiti in competizione quando somministrati tramite vie “sistemiche” (orali, rettali, intramuscolari o endovenose).[https://www.wada-ama.org/] La somministrazione tramite tutte le altre vie (incluse le iniezioni intra-articolari e altre periarticolari) è considerata somministrazione locale e non è proibita in competizione. La somministrazione di Glucocorticoidi tramite qualsiasi via non è proibita fuori competizione (OOC).
Indipendentemente dalla sostanza specifica del Glucocorticoidi e dalle sue singole caratteristiche farmacologiche, un presunto riscontro analitico avverso (AAF) viene segnalato dai laboratori accreditati WADA quando i livelli urinari dei campioni in gara superano un livello di segnalazione di 30ng/mL. La farmacocinetica dei Glucocorticoidi è complessa e influenzata dalla formulazione, dal tipo di esterificazione e sale, dalla via di somministrazione, dal sito e dal metodo di somministrazione. Di conseguenza, mentre il limite di segnalazione del laboratorio può dimostrare la presenza di un Glucocorticoidi , non può necessariamente indicare se la somministrazione è avvenuta in gara o OOC o se è probabile che abbia un effetto farmacologico o ergogenico. Qualsiasi medico o atleta non sarà sicuro di quando interrompere l’uso di GC sistemici prima del periodo in gara per evitare di superare il limite di segnalazione. Per complicare ulteriormente il quadro farmacocinetico, le iniezioni intra-articolari possono dare origine a livelli sistemici e i medici possono inavvertitamente caratterizzare erroneamente il sito di iniezione in assenza di guida radiologica o ecografica. La definizione di limiti di segnalazione specifici per sostanza è un’area di discussione e ricerca attiva tra gli esperti nominati dalla WADA e va oltre lo scopo del presente documento.
Glucocorticoidi sistemici e performance
Alcuni atleti hanno indubbiamente tentato di sfruttare i presunti effetti di miglioramento delle prestazioni dei Glucocorticoidi sistemici che ritengono benefici nella loro particolare disciplina sportiva. Tuttavia, i meccanismi complessi e pleiotropici dell’azione dei Glucocorticoidi suggeriscono che questi farmaci sono uno strumento poco maneggevole per l’atleta che cerca di ottenere un vantaggio nelle prestazioni e sono considerati una componente meno popolare dei regimi di doping rispetto al passato.[ https://cyclingtips.com/] Alcuni pazienti e atleti hanno riferito di aver sperimentato euforia dopo la somministrazione sistemica.[ http://www.cyclingnews.com] Tuttavia, le prove scientifiche a supporto dell’euforia misurabile nelle popolazioni cliniche sono ambigue e l’interpretazione dei dati è complicata dall’associazione del dolore cronico confondente.[https://bjsm.bmj.com/]
Sembrerà starno, ma non vi è alcuna prova incontrovertibile di effetti di miglioramento delle prestazioni derivanti dall’uso a breve termine di Glucocorticoidi sistemici.[https://bjsm.bmj.com/] Esistono studi randomizzati in doppio cieco cross-over che suggeriscono che gli atleti possono sfruttare cicli di Glucococrticoidi orali ad alto dosaggio della durata di una settimana per migliorare le loro prestazioni di esercizio di intensità submassimale per brevi periodi di tempo.[https://bjsm.bmj.com/] Questi dosaggi sarebbero facilmente rilevati durante i test antidoping, se assunti in gara. Il meccanismo preciso di questo effetto non è chiaro, ma si suggerisce che derivi da una combinazione di effetti sul metabolismo energetico, sui muscoli, sull’infiammazione e sul sistema nervoso. Questo effetto del farmaco è stato dimostrato in uno studio su atleti maschi il cui allenamento era strettamente periodizzato insieme all’uso di Glucocorticoidi orali.[https://bjsm.bmj.com/] Sfruttare questo tipo di regime di miglioramento delle prestazioni evitando efficacemente l’insufficienza surrenalica e il rilevamento tramite controlli antidoping standard in gara richiederebbe una meticolosa supervisione medica. Potrebbe anche richiedere una manipolazione farmacologica più complessa ed esotica dell’asse ipotalamo-ipofisi-surrene rispetto a quella offerta dai Glucocorticoidi prescritti.[https://bjsm.bmj.com/]
Atleti e dottori hanno descritto metodi inappropriati con cui l’uso sistemico di Glucocorticoidi, un’alimentazione limitata e un allenamento a bassa intensità potrebbero essere combinati OOC per perdere peso e preservare la massa muscolare.[ https://www.nytimes.com/]Tuttavia, date le funzioni cataboliche proteiche ampiamente riconosciute dei Glucocorticoidi,[https://bjsm.bmj.com/] questo meccanismo di doping rimane speculativo e controverso. Inoltre, l’efficacia potrebbe dipendere dall’uso di Glucocorticoidi come parte di un cocktail complesso che include altri ormoni proibiti ma scarsamente rilevati come l’insulina.[https://bjsm.bmj.com/]
Schema esemplificativo sull’uso di Glucocorticoidi sistemici e perdita di peso/ricomposizione corporea:la base teorica sulla quale si sostiene la suddetta pratica farmacologica è il ciclo di feedback negativo dell’Asse HPA per via d’uso di Glucocorticoidi esogeni. Tale pratica dovrebbe portare a 1) soppressione del rilascio di —> CRH>ACTH>Cortisone<>Cortisolo con consequenziale prevenzione di 2) aumento della fame/appetito con il procedere del regime ipocalorico 3) prevenzione del aumento del catabolismo muscolare 4) prevenzione di un aumento della ritenzione idrica e 5) prevenzione dell’alterazione del metabolismo lipidico correlato ad un incremento significativo del Cortisolo. Da notare che il momento della somministrazione del Glucocorticoide esogeno può influenzare il grado di soppressione surrenalica. Per esempio, il Prednisone in una dose di 5mg somministrato la sera prima di coricarsi e 2,5mg al mattino produrrà una soppressione dell’Asse HPA più marcata rispetto a 2,5mg la sera e 5mg al mattino. 5mg è un dosaggio basso e generalmente non è promotore di insonnia sebbene in soggetti sensibili può manifestarsi. Il dosaggio comunemente utilizzato varia da 15 a 25mg/die diviso in due dosi dopo i pasti. Tale pratica comunemente è parte di protocolli PEDs più complessi e contenenti uno o più agenti anabolizzanti.
Recenti resoconti sulla presunta potenza dei Glucocorticoidi sistemici provengono da atleti che hanno anche confessato l’uso concomitante di altri metodi e sostanze per migliorare le prestazioni, tra cui agenti anabolizzanti come il testosterone.[https://bjsm.bmj.com/] Tali regimi di Glucocorticoidi potrebbero avere rilevanza solo in un piccolo sottoinsieme di discipline sportive, come nelle ripide tappe di montagna dei Grandi Giri del ciclismo, dove gli atleti potrebbero essere disposti ad accettare compromessi nei loro regimi di allenamento o potenza assoluta in uscita nel perseguimento di un rapporto potenza/peso superiore. L’uso di OOC richiederebbe comunque una continuazione prolungata dell’uso di Glucocorticoidi nel periodo di gara per evitare l’insufficienza surrenalica dovuta a meccanismi di feedback. L’uso prolungato di Glucocorticoidi comporta rischi medici ben noti, alcuni dei quali potrebbero ridurre in modo permanente le prestazioni atletiche.[https://bjsm.bmj.com/]
Rischi per la salute, eventi avversi ed effetti negativi sulle prestazioni
Il trattamento con Glucocorticoidi per molte condizioni ha una lunga storia e un profilo di sicurezza ragionevole. Dosi elevate o uso cronico di Glucocorticoidi sistemici presentano un certo rischio per la salute dell’atleta. Un esame attento, una diagnosi e una deliberazione da parte del medico sono fondamentali e i benefici del trattamento devono essere soppesati rispetto ai potenziali rischi ed effetti avversi. L’uso potenziale per migliorare le prestazioni, descritto sopra e ritenuto limitato a contesti sportivi specifici con uso di GC ad alto dosaggio, è anche potenzialmente associato a rischi significativi per la salute di un atleta.
Gli eventi avversi con associazioni causali ben consolidate all’uso clinicamente appropriato di GC toccano praticamente ogni sistema umano, vanno da esiti negativi sulla salute acuti a cronici e includono insufficienza surrenalica, immunodeficienza, osteoporosi, atrofia muscolare, cedimento di tendini/fasce, necrosi avascolare della testa femorale, vari squilibri elettrolitici, nutrizionali e metabolici, glaucoma e cataratta. Forse perché i GC sono farmaci così comuni e clinicamente versatili, alcuni medici potrebbero sopravvalutare il loro valore terapeutico e sottostimare la gravità degli eventi avversi associati.[https://bjsm.bmj.com/] Anche una singola iniezione intra-articolare potrebbe causare un’insufficienza surrenalica clinicamente significativa che porta a malessere, squilibrio elettrolitico e immunosoppressione per diverse settimane.[https://bjsm.bmj.com/]
È importante sottolineare che l’eziologia di questi sintomi potrebbe non essere riconosciuta dall’atleta e dal personale medico, in particolare in un contesto sportivo in cui gli atleti si allenano ad alta intensità e i sintomi possono mascherarsi da affaticamento correlato al sovrallenamento. Inoltre, un atleta che subisce un trauma o un infortunio grave potrebbe essere a maggior rischio di crisi surrenalica a causa della soppressione ipotalamo-ipofisi-surrene dovuta al precedente utilizzo di GC. Ciò potrebbe essere particolarmente problematico se l’atleta non rivela questo precedente utilizzo.
Sia l’efficacia che il potenziale danno delle iniezioni intra-articolari sono ampiamente dibattuti. Le prove di un recente studio prospettico controllato con placebo su pazienti con osteoartrite hanno suggerito che frequenti iniezioni di triamcinolone al ginocchio, somministrate secondo un programma prestabilito, non sono riuscite a gestire efficacemente il dolore a lungo termine e hanno portato a una riduzione statisticamente significativa dello spessore della cartilagine.[https://bjsm.bmj.com/]Tuttavia, le raccomandazioni della società medica, così come una meta-analisi completa, supportano l’efficacia e la sicurezza dello stesso intervento,[https://bjsm.bmj.com/l] suggerendo fortemente che un uso giudizioso di iniezioni intra-articolari in pazienti e circostanze appropriate può produrre risultati positivi. Vi è una mancanza di prove pubblicate sulla sicurezza o il danno dell’uso di GC intra-articolari nelle popolazioni di atleti e sono urgentemente necessarie ulteriori ricerche a causa dell’uso onnipresente di GC intra-articolari.
Politiche per garantire l’uso appropriato dei GC
Nonostante le preoccupazioni di un possibile abuso per un vantaggio competitivo o potenziali effetti dannosi sulla salute degli atleti, i GC sono ampiamente utilizzati nello sport per legittime ragioni terapeutiche. Considerando che l’elenco è armonizzato in tutti gli sport, dal tiro con l’arco al wakeboard, il doping con i GC non è un problema laddove i presunti benefici dell’uso di GC ad alto dosaggio (potenza prolungata a intensità di esercizio submassimali o gestione aggressiva del peso catabolico) difficilmente miglioreranno le prestazioni. Pertanto, un AAF per i GC non sarebbe probabilmente associato a nessun intento di doping. L’uso di GC sistemici in molti sport deve essere considerato sotto una luce diversa rispetto agli sport ad alto rischio come il ciclismo, dove l’abuso è ben documentato e le prove scientifiche forniscono un certo supporto.
Consapevoli delle sfide specifiche poste dall’uso di GC nello sport, le organizzazioni sportive e antidoping hanno introdotto politiche innovative e stanno rafforzando le normative esistenti per affrontare l’uso terapeutico ragionevole dei GC.
Conclusioni su Glucocorticoidi e prestazioni sportive:
Da recenti review, sappiamo che l’uso dei Glucocorticoidi sistemici può migliorare la prestazione fisica massima rispetto al placebo (SDM 0,300, 95% CI 0,080-0,520). In una recente review [https://www.frontiersin.org/], l’SDM per i 13 confronti inclusi non era eterogeneo (I2 = 35%, p = 0,099). L’analisi di sensibilità escludendo i due studi con alto rischio di bias ha mostrato un effetto simile (SDM 0,349, 95% CI 0,071-0,626). Con la meta-regressione si è scoperto che la durata del trattamento, la via di somministrazione e il tipo di esercizio non hanno influenzato (p > 0,124) l’SDM. Nell’analisi stratificata il trattamento prolungato e l’ingestione orale hanno migliorato la prestazione fisica (p = 0,003). Il trattamento acuto e l’inalazione non hanno avuto alcun effetto sulla prestazione fisica (p > 0,564), l’analisi di sensibilità con studi ad alto rischio di bias rimossi o solo un trattamento per gruppo di controllo, ha mostrato un effetto simile all’analisi completa con SDM 0,334, 95% CI 0,075-0,592 e SDM 0,296 0,059-0,532, rispettivamente. L’analisi di sensibilità escludendo i sei confronti con meno di 10 coppie di dati non ha indicato alcun effetto dei Glucocorticoidi sulla prestazione fisica (p = 0,070). I Glucocorticoidi hanno migliorato la prestazione aerobica (SDM 0,348, 95% CI 0,129-0,567). Tre confronti hanno testato l’effetto dei Glucocorticoidi sulla prestazione anaerobica massima e la meta-analisi dei confronti non ha mostrato alcun effetto (p = 0,573) sulla prestazione fisica. L’effetto è rimasto non statisticamente significativo dopo aver incluso i due studi che misuravano la prestazione anaerobica all’interno di un test di prestazione aerobica (p = 0,491) e quando tutti i risultati della prestazione anaerobica negli studi inclusi (anche più risultati dello stesso studio) sono stati meta-analizzati (p = 0,177). Non è stato trovato alcun effetto dei Glucocorticoidi sulla spesa energetica durante la prestazione submassimale (SDM 0,-332, 95% CI −0,785 a 0,121).
In una review narrativa del 2016, Collomp et al. hanno mirato a riassumere le attuali conoscenze sugli effetti ergogenici dei glucocorticoidi negli esseri umani. Hanno riferito che un effetto ergogenico (sull’esercizio di endurance) dei Glucocorticoidi sistemici a breve termine è stato chiaramente dimostrato e che gli effetti a breve termine (4,5 giorni) e a lungo termine (4 settimane) dell’assunzione di Glucocorticoidi non hanno avuto alcun effetto sul VO2-max o sulla potenza massima in uscita durante protocolli di esercizio graduati. Hanno anche il test sul campo come categoria di test delle prestazioni, ma fanno riferimento solo allo studio di Casuso et al. che riporta un miglioramento delle prestazioni nella corsa a navetta, ma nessun cambiamento nelle prestazioni nello sprint. I risultati successivi supportano e rafforzano la conclusione di Collomp et al. ( per quanto riguarda l’esercizio di endurance poiché scopriamo anche che il trattamento prolungato con glucocorticoidi migliora le prestazioni aerobiche. Tuttavia, a differenza di Collomp et al. nelle ultime review sono stati inclusi sia test di esercizio graduati che test sul campo (della durata di 1 minuto o più) nella definizione di prestazione aerobica e quindi, più studi che hanno aggiunto potenza statistica alla analisi. Non è stato trovato alcun effetto dei Glucocorticoidi sulla prestazione anaerobica quando è stato analizzato secondo il protocollo di studio, tuttavia sono stati inclusi solo tre studi che testavano la prestazione anaerobica. Per aumentare la potenza statistica, è stato anche incluso la prestazione anaerobica all’interno di test aerobici e più test anaerobici dallo stesso studio, ma ancora non era evidente alcun effetto dei glucocorticoidi. Questo approccio può diminuire la validità dell’analisi poiché la prestazione anaerobica all’interno di test aerobici può testare altre abilità rispetto ai test anaerobici e la meta-analisi di più risultati dagli stessi soggetti e l’intervento non è raccomandato dal Cochrane Handbook. Collomp et al. concludono che non è chiaro se i Glucocorticoidi migliorino la prestazione durante l’esercizio breve-intenso. Questa conclusione è ancora valida poiché solo pochi studi hanno indagato l’effetto dei glucocorticoidi sull’esercizio anaerobico/breve intenso. Tuttavia, quando meta-analizziamo tutte le prove disponibili, sembra che i Glucocorticoidi non migliorino le prestazioni anaerobiche. I Glucocorticoidi non hanno influenzato le prestazioni submassimali aumentando l’energia totale spesa e/o il VO2max a un carico fisso, ma questa conclusione dovrebbe anche essere interferita con cationi poiché l’analisi include solo 35 soggetti, il che fornisce una potenza statistica limitata.
L’uso di Glucocorticoidi nello sport è una questione altamente complessa a causa del loro uso diffuso in medicina, delle numerose formulazioni e vie di somministrazione con farmacocinetica variabile, effetti negativi sulla salute e potenziali associazioni di doping.
In definitiva, da quanto recentemente emerso attraverso il riassunto delle migliori prove scientifiche disponibili, i Glucocorticoidi migliorano le prestazioni aerobiche e massime, ma non influenzano le prestazioni anaerobiche nei soggetti sani.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Pelt AC (2011). Glucocorticoids: effects, action mechanisms, and therapeutic uses. Hauppauge, N.Y.: Nova Science. ISBN978-1617287589.[page needed]
Ralph CR, Tilbrook AJ (February 2016). “INVITED REVIEW: The usefulness of measuring glucocorticoids for assessing animal welfare”. Journal of Animal Science. 94 (2): 457–470. doi:10.2527/jas.2015-9645. PMID27065116.
Collomp K, Arlettaz A, Buisson C, Lecoq AM, Mongongu C. Glucocorticoid administration in athletes: performance, metabolism and detection. Steroids. (2016) 115:193–202. doi: 10.1016/j.steroids.2016.09.008 PubMed Abstract | CrossRef Full Text | Google Scholar
Fain JN, Cheema P, Tichansky DS, Madan AK. Stimulation of human omental adipose tissue lipolysis by growth hormone plus dexamethasone. Mol Cell Endocrinol. (2008) 295(1):101–5. doi: 10.1016/j.mce.2008.05.014PubMed Abstract | CrossRef Full Text | Google Scholar
Thomasson R, Rieth N, Jollin L, Amiot V, Lasne F, Collomp K. Short-term glucocorticoid intake and metabolic responses during long-lasting exercise. Horm Metab Res. (2011) 43(3):216–22. doi: 10.1055/s-0030-1269919 PubMed Abstract | CrossRef Full Text | Google Scholar
Duclos M, Guinot M, Le Bouc Y, Cortisol GH. Odd and controversial ideas. Appl Physiol Nutr Metab. (2007) 32(5):895–903. doi: 10.1139/h07-064PubMed Abstract | CrossRef Full Text | Google Scholar
Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. (2000) 21(1):55–89. doi: 10.1210/edrv.21.1.0389PubMed Abstract | CrossRef Full Text | Google Scholar
Duclos M. Evidence on ergogenic action of glucocorticoids as a doping agent risk. Phys Sportsmed. (2010) 38(3):121–7. doi: 10.3810/psm.2010.10.1817PubMed Abstract | CrossRef Full Text | Google Scholar
Strehl C, Bijlsma JWJ, de Wit M, Boers M, Caeyers N, Cutolo M, et al. Defining conditions where long-term glucocorticoid treatment has an acceptably low level of harm to facilitate implementation of existing recommendations: viewpoints from an EULAR task force. Ann Rheum Dis. (2016) 75(6):952–7. doi: 10.1136/annrheumdis-2015-20891PubMed Abstract | CrossRef Full Text | Google Scholar
Ventura R, Daley-Yates P, Mazzoni I, Collomp K, Saugy M, Buttgereit F, et al. A novel approach to improve detection of glucocorticoid doping in sport with new guidance for physicians prescribing for athletes. Br J Sports Med. (2021) 55(11):631–42. doi: 10.1136/bjsports-2020-103512
Zhou M, Gomez-Sanchez CE (July 1993). “Cloning and expression of a rat cytochrome P-450 11 beta-hydroxylase/aldosterone synthase (CYP11B2) cDNA variant”. Biochem. Biophys. Res. Commun. 194 (1): 112–7. doi:10.1006/bbrc.1993.1792. PMID8333830.
Lefebvre H, Cartier D, Duparc C, et al. (March 2002). “Characterization of serotonin(4) receptors in adrenocortical aldosterone-producing adenomas: in vivo and in vitro studies”. J. Clin. Endocrinol. Metab. 87 (3): 1211–6. doi:10.1210/jcem.87.3.8327. PMID11889190.
Lupien SJ, McEwen BS, Gunnar MR, Heim C (Jun 2009). “Effects of stress throughout the lifespan on the brain, behaviour and cognition”. Nature Reviews. Neuroscience. 10 (6): 434–445. doi:10.1038/nrn2639. PMID19401723. S2CID205504945.
Belanoff JK, Gross K, Yager A, Schatzberg AF (2001). “Corticosteroids and cognition”. Journal of Psychiatric Research. 35 (3): 127–145. doi:10.1016/S0022-3956(01)00018-8. PMID11461709.
Lupien SJ, Maheu F, Tu M, Fiocco A, Schramek TE (Dec 2007). “The effects of stress and stress hormones on human cognition: Implications for the field of brain and cognition”. Brain and Cognition. 65 (3): 209–237. doi:10.1016/j.bandc.2007.02.007. PMID17466428. S2CID5778988.
Wolkowitz OM, Lupien SJ, Bigler ED (Jun 2007). “The ‘steroid dementia syndrome’: a possible model of human glucocorticoid neurotoxicity”. Neurocase. 13 (3): 189–200. doi:10.1080/13554790701475468. PMID17786779. S2CID39340010.
Liu C, Chen Y, Kang Y, Ni Z, Xiu H, Guan J, et al. (Oct 2011). “Glucocorticoids improve renal responsiveness to atrial natriuretic peptide by up-regulating natriuretic peptide receptor-A expression in the renal inner medullary collecting duct in decompensated heart failure”. The Journal of Pharmacology and Experimental Therapeutics. 339 (1): 203–209. doi:10.1124/jpet.111.184796. PMID21737535. S2CID1892149.
Newton R, Holden NS (Oct 2007). “Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor?”. Molecular Pharmacology. 72 (4): 799–809. doi:10.1124/mol.107.038794. PMID17622575. S2CID52803631.
Rado JP, Blumenfeld G, Hammer S (Nov 1959). “The effect of prednisone and 6-methylprednisolone on mercurial diuresis in patients with refractory cardiac edema”. The American Journal of the Medical Sciences. 238 (5): 542–551. doi:10.1097/00000441-195911000-00003. PMID14435747. S2CID38687480.
Riemer AD (Apr 1958). “Application of the newer corticosteroids to augment diuresis in congestive heart failure”. The American Journal of Cardiology. 1 (4): 488–496. doi:10.1016/0002-9149(58)90120-6. PMID13520608.
Newman DA (Feb 1959). “Reversal of intractable cardiac edema with prednisone”. New York State Journal of Medicine. 59 (4): 625–633. PMID13632954.
Massari F, Mastropasqua F, Iacoviello M, Nuzzolese V, Torres D, Parrinello G (Mar 2012). “The glucocorticoid in acute decompensated heart failure: Dr Jekyll or Mr Hyde?”. The American Journal of Emergency Medicine. 30 (3): 517.e5–10. doi:10.1016/j.ajem.2011.01.023. PMID21406321.
Il collagene (o collageno) è la principale proteina fibrosa del tessuto connettivo negli animali. È la proteina più abbondante nei mammiferi (circa il 25% della massa proteica totale), rappresentando nell’uomo circa il 6% del peso corporeo.
Il collagene, quindi, è il componente strutturale primario di tutti i tessuti connettivi (principalmente collagene di tipo I per ossa, tendini e legamenti; e principalmente collagene di tipo II per la cartilagine). L’attività del collagene di tipo III riflette la crescita e il turnover dei tessuti molli; è ampiamente distribuito nelle fibrille interstiziali dei tessuti molli, compresi tendini e legamenti, fascia, nonché nella matrice extracellulare (ECM) e nei suoi strati (ad esempio, l’endomisio) che avvolgono le parti costitutive dei gruppi muscolari e che contengono vari fattori di crescita coinvolti nella funzione e nella crescita del muscolo scheletrico.
Il metabolismo del collagene comprende i processi biochimici che regolano la sintesi, la degradazione e il ricambio dei tessuti molli che sono costituiti principalmente da collagene. I tessuti connettivi del nostro corpo sono in uno stato costante di equilibrio e flusso. Il metabolismo del collagene mantiene l’integrità strutturale e la funzione di articolazioni, ossa, tendini e legamenti del corpo. Gli enzimi e le vie di segnalazione regolano questi processi di sintesi, scomposizione e ricambio.
Il procollagene, la proteina madre del collagene, è sintetizzata e secreta dai fibroblasti. Le molecole di procollagene sono costituite da 3 filamenti proteici disposti a tripla elica. Il procollagene lascia la cellula con estensioni protettive alle estremità per prevenire la formazione prematura di collagene. La scissione dei prolungamenti tramite enzimi porta alla formazione di collagene attivo che si allinea con altre molecole di collagene.
Punto chiave: La misurazione di questi enzimi (cioè dei marcatori) fornisce un’indicazione del metabolismo del collagene. Un aumento o una diminuzione dei livelli di alcuni enzimi indica la sintesi netta di collagene (ad esempio, di tipo I), mentre una diminuzione o un aumento dei livelli di alcuni enzimi indica la degradazione netta di collagene (ad esempio, di tipo I).
Una microfibrilla è un’unità di filamenti di collagene disposti in parallelo. Sono le subunità delle fibre di collagene. Le fibre sono disposte in fasci. I legami incrociati tra le molecole di collagene adiacenti nei fasci di collagene sono legami chimici forti che garantiscono l’integrità e un robusto reticolo di tessuti connettivi che supportano lo scheletro nella locomozione.
Il collagene di tipo I è particolarmente importante per le modifiche del contenuto minerale osseo e della densità minerale ossea (BMC/BMD).
Il collagene di tipo III è particolarmente importante per i cambiamenti nella ECM, nei tendini, nei legamenti e nei tessuti connettivi:
Trasmissione della forza dal sarcomero all’osso (aumento della forza)
Recupero da lesioni muscoloscheletriche, in particolare quelle che coinvolgono la matrice di collagene.
Prevenzione di lesioni muscoloscheletriche da uso eccessivo o acute (aumentando il rapporto forza-fatica).
In generale gli AAS influenzano il metabolismo del collagene. AAS sovrafisiologici:
↑ PIIINP sierica [in maniera dose-dipendente]
↑ urina HP:LP – Dosi elevate di AAS aumentano il metabolismo del collagene dei tessuti molli, senza variazioni nel riassorbimento osseo. [1].
Cessazione (“cycling-off”) d’uso degli AAS:
↓ ICTP sierico [in funzione del tempo]. [1].
Marker del metabolismo del collagene:
Tendine ( marker metabolici del collagene)
Il propeptide N-terminale del procollagene di tipo III (PIIINP) è un marker della biosintesi delle fibrille interstiziali nei tessuti molli. Il PIIINP è stimolato dall’allenamento pliometrico nei tendini e dagli AAS in generale, dove la rigidità va a vantaggio della velocità, ma l’aumento della forza muscolare deve compensare l’aumento del rischio di strappi muscolari dovuto all’eccessiva rigidità dei tendini.
-Aumenta in modo dose-dipendente con gli AAS a concentrazione sovrafisiologica.
Ossa e tendini (“marker metabolici del collagene”)
Procollagene di tipo I C-terminale propeptide (PICP): rilasciato in circolo dagli osteoblasti proliferanti durante la biosintesi del collagene, è quindi in gran parte un biomarcatore della formazione di collagene osseo, sebbene vi sia anche un certo contributo da parte del collagene di tipo I nei tessuti molli. [2].
Telopeptide reticolato del collagene di tipo I (ICTP): rilasciato in circolo durante la fase osteoclastica della modellazione e del rimodellamento osseo (disgregazione dell’osso/collagene). [2].
-Diminuisce con la sospensione di AAS sovrafisiologici (“cycling-off”) in modo dipendente dal tempo.
Dpyr: marcatore urinario della degradazione del collagene di tipo I. [2].
Sia l’ICTP che il Dpyr costituiscono legami incrociati piridinolinici formati nel collagene maturo di tipo I e le loro concentrazioni nel siero e nelle urine riflettono il riassorbimento osseo. [2].
Ossa (“marker metabolici del collagene”)
Fosfatasi alcalina ossea (ALP): proteina presente nelle cellule ossee della placca di crescita epifisaria e negli osteoblasti maturi. È irrilevante per le dimensioni o la forza muscolare. [2].
HP (PYD): idrossilisilpiridinolina; riflette il turnover dei collageni di tipo I (osso), II, III e IX; presente in tendini, cartilagini, ossa, pareti dei vasi e dentina. [2].
LP (DPD): lisilpiridinolina; riflette il turnover del collagene di tipo I (osso); presente nell’osso e nella dentina.
Esiste un declino associato all’età di HP (↓), LP (↓) e del rapporto HP/LP (↓).
HP:LP (urina) esclude il metabolismo cutaneo (utile). [2].
Idrossiprolina
L’HP e l’LP urinari sono marcatori potenzialmente più utili del catabolismo delle fibre di collagene dei tessuti scheletrici rispetto all’idrossiprolina urinaria. Quest’ultima ha un profilo di specificità inferiore poiché si trova in tutti i tipi di collagene di tutti i tessuti connettivi (compresa la pelle). Inoltre, può anche essere rilasciata dalle molecole di collagene prima della loro incorporazione nelle fibrille e una grande percentuale di idrossiprolina viene metabolizzata nel fegato, eludendo così l’analisi quantitativa del riassorbimento del collagene maturo mediante misurazioni urinarie. [3].
Un rapporto HP:LP più basso può riflettere una maggiore proporzione relativa di riassorbimento osseo rispetto al turnover del collagene e della cartilagine, poiché il collagene osseo è la fonte primaria di LP, mentre l’HP riflette i tessuti molli in generale (eccetto la pelle)… suggerisce che anche il riassorbimento del collagene di tipo III (più HP rispetto al tipo I) diminuisce con l’età. [3].
-Aumenta con gli AAS sovrafisiologici.
Il picco di massa ossea e di HP:LP si verifica all’età di 27 anni. [3].
Ossa:
Cellule ossee
Osteoprogenitori: Cellule osteogeniche che si sviluppano in osteoblasti.
Osteoblasti: Formano l’osso; producono ECM ossea e mineralizzano l’osso (mononucleati).
Osteociti: Cellule ossee mature; secernono enzimi per mantenere l’osso.
La formazione ossea osteoblastica è associata alla deposizione di collagene di tipo I, seguita dalla mineralizzazione e dalla maturazione, durante le quali si formano legami incrociati stabili tra le fibrille di collagene. [4].
Osteocalcina (siero): Riflette la formazione ossea (così come il procollagene di tipo I [siero]).
Deossipiridinolina (urina): Riflette il riassorbimento osseo. [4].
Tendini e legamenti:
Il tendine è un tessuto connettivo che collega l’osso al muscolo, mentre il legamento è un tessuto connettivo che collega l’osso all’osso. In entrambi, circa ¾ del peso secco è costituito da collagene: la maggior parte è di tipo I: 60% (tendine) e fino all’85% (legamento).
La struttura relativamente (quasi totalmente) avascolare e collagena di legamenti e tendini limita il loro potenziale rigenerativo, con conseguenti complicazioni mediche sostanziali, che spesso rendono necessario un intervento chirurgico dopo una lesione traumatica.
I tendini e i legamenti maturi contengono relativamente poche cellule. Il numero ridotto di cellule metabolicamente attive comporta un fabbisogno di ↓O₂ e di nutrienti. I legamenti contengono fibre di elastina e collagene.
Le proprietà meccaniche di tendini e legamenti sono funzione di:
Densità delle fibre di collagene
Diametro
Orientamento e
Reticolazione -Legami incrociati enzimatici, formati dall’ossido di lisile (LOX) -Legami incrociati non enzimatici attraverso gli AGE (advanced glycation end-products), formati da una reazione di Maillard senza enzimi specifici tra uno zucchero e un amminoacido. -Entrambi i legami incrociati aumentano la rigidità di tendini e legamenti. [5].
Adattamenti dell’allenamento
Il metabolismo del tendine è molto più lento di quello del muscolo a causa della sua ridotta vascolarizzazione e circolazione, e l’aumento del flusso sanguigno al muscolo scheletrico attraverso l’esercizio fisico non è parallelo alla stessa perfusione del flusso nel tendine. [6].
L’ipertrofia muscolare è correlata a un aumento del numero e delle dimensioni dei fibroblasti, con conseguente aumento dell’apporto totale di collagene. L’attivazione dei fibroblasti e la successiva crescita del tessuto connettivo sono i prerequisiti dell’ipertrofia [7], in modo che il contenuto di collagene sia mantenuto in proporzione alla massa muscolare.
La rigidità del tendine si riferisce alla trasmissione della forza per unità di sforzo, o allungamento del tendine. L’aumento della rigidità del legamento è una buona cosa (✓), poiché la rigidità è utile per mantenere la stabilità dell’articolazione e il rischio di lesioni. Al contrario, poiché il tendine collega l’osso rigido al muscolo cedevole, un tendine più rigido non è sempre vantaggioso:
In termini di prestazioni: ↑La rigidità (tendine) trasmette più velocemente le forze muscolari all’osso, con conseguente ↑ prestazione; tuttavia, questo interesse deve essere bilanciato dal potenziale di concentrazione delle deformazioni all’interno del muscolo:
La maggiore deformazione (“stiramento”) prodotta in un determinato movimento si concentra nel muscolo collegato a un tendine rigido ⇒ contrazione isometrica piuttosto che nel tendine che si allunga in modo flessibile mentre il muscolo si contrae.
Un tendine rigido non si allunga; piuttosto, è costretto ad allungarsi durante la contrazione (eccentrica), pertanto un muscolo collegato a un tendine rigido subisce un carico eccentrico maggiore per un determinato movimento e presenta un rischio maggiore di lesioni. [5].
Effetti degli androgeni sul tendine:
Tabella che descrive le prove degli effetti meccanici, strutturali e biologici degli AAS sul tendine. [9].
Il risultato di questa tabella mostra che gli effetti degli AAS sul tendine, e soprattutto sull’unità muscolo-tendinea, non si prestano a una descrizione univoca o a una conclusione univoca, ma presentano sfumature dovute all’eterogeneità dei dati. Gli AAS migliorano e ostacolano diversi elementi della struttura e della funzione del tendine.
In sintesi, le conclusioni pratiche che si possono trarre dalla letteratura sugli effetti degli AAS sul tendine sono le seguenti:
Gli effetti biomeccanici potenzialmente deleteri degli AAS possono essere transitori.
Gli AAS somministrati per via sistemica e locale possono avere effetti simili sui tendini.
Esistono notevoli lacune nelle conoscenze relative a: -Lesioni/patologia del tendine -Risposta alla dose -Risposta al farmaco (la maggior parte degli studi utilizza il nandrolone; alcuni il metandienone) -Cosomministrazione -Tempistica e -Popolazione di risposta (differenze legate al sesso e all’età).
Effetti specifici sui tessuti: -È probabile che gli AAS influenzino in modo diverso il metabolismo dei tendini, dei muscoli e della fibrocartilagine (sintesi/degradazione netta), ma finora non sono stati condotti studi per caratterizzare queste differenze. [9].
Articolazioni:
Le articolazioni del corpo sono i punti in cui le ossa si incontrano (articolazione) e che consentono la locomozione umana (i muscoli tirano le ossa sulle articolazioni per muovere il corpo). Le articolazioni sono costituite da ossa, tendini (che collegano le ossa ai muscoli) e legamenti (che collegano le ossa alle ossa). È possibile classificare le articolazioni secondo vari schemi (ad esempio, funzionale, strutturale). Questo articolo si concentrerà sulle articolazioni sinoviali dell’organismo, che comprendono l’anca, il ginocchio, la spalla e il gomito. Oltre a essere composte da ossa, tendini e legamenti, queste articolazioni contengono cellule sinoviali, un tipo di cellula che contiene una membrana di rivestimento che produce il liquido sinoviale. Il liquido sinoviale lubrifica e nutre l’articolazione, riduce l’attrito e fornisce ammortizzazione tra le superfici articolari. È importante notare che questa lubrificazione e questo nutrimento dipendono dalla produzione di proteine e fattori di crescita.
Gli effetti degli AAS sulle articolazioni non sono descritti in modo esaustivo, ma esistono studi sui singoli composti che verranno esaminati in seguito per approfondire i particolari AAS e i loro effetti sulle articolazioni. I potenziali meccanismi con cui gli AAS influenzano le articolazioni riguardano in generale gli effetti sul metabolismo, sul ricambio, sulla scomposizione e sulla sintesi del collagene, gli effetti sul C1-INH e la funzione delle articolazioni sinoviali.
C1-INH:
Inibitore della C1-esterasi; SERPING1
Struttura molecolare del C1-INH
Il C1-INH è un inibitore della proteasi multi-serina che controlla diverse vie catalitiche, tra cui l’attivazione dei componenti classici.
Gli androgeni attenuati – AAS che possiedono una potenza androgena relativamente ridotta – qui discussi, l’Oxandrolone e lo Stanozololo, entrambi 17AA, inducono la produzione intrinseca di C1-INH e il ↑catabolismo (cioè la scomposizione) della bradichinina.
La bradichinina, attraverso la sua azione sul B₂R, media la vasodilatazione e aumenta la permeabilità con conseguente angioedema. L’angioedema ereditario (HAE), per estensione, deriva da ↓C1-INH. L’HAE è una condizione che può essere trattata con androgeni attenuati.
Quando il C1-INH è ridotto, si verifica una permeabilità vascolare (sottocutanea e sottomucosa) (“angioedema”) dovuta alla ↑bradichinina (che il C1-INH attenua) [a causa degli effetti sul sistema di contatto classico e sull’attivazione del complemento che esulano dallo scopo di questo articolo]. [10].
Gli androgeni regolano l’espressione genica della C1-INH e l’aminopeptidasi P plasmatica (che catabolizza le chinine) (55, 56). [10].
Gli androgeni attenuati (17AA) aumentano in modo più potente la produzione epatica di C1-INH [23] per azione diretta a livello epatico piuttosto che per azione dell’AR di per sé ⇒ ↑C1-INH e C4 (a causa dell’inversione dei livelli secondariamente depressi di C4). [11].
I complessi C1/C1-INH si formano quando C1-INH si combina e rimuove C1r e C1s dal C1 attivato e questi complessi – rappresentativi dell’attivazione della via del complemento classica – sono associati a condizioni artritiche e reumatologiche. [12]. Ciò può contribuire alla reputazione di Stanozolol, in particolare, di causare dolori articolari.
Punto chiave: L'”angioedema acquisito senza focolai” può essere causato da farmaci, di cui gli ACE-inibitori (nello 0,1-2,2% dei consumatori, più numerosi negli africani) sono i responsabili più comuni. L’ACE è necessario per la degradazione della bradichinina e la sua inibizione può provocare un accumulo di bradichinina che causa l’angioedema. Gli androgeni possono in qualche misura sopprimere l’angioedema grazie ai loro effetti sulla C1-INH. In un articolo di prossima pubblicazione di questo autore si discutono i rischi della diffusione di farmaci antipertensivi, soprattutto ACE inibitori e ARB, per i bodybuilder sani che fanno uso di AAS in assenza di ipertensione cronica, e si affronta la questione particolare (e apparentemente controversa) della diminuzione (blunting) dell’ipertrofia indotta dagli ACE inibitori.
Molecole:
Nandrolone
Scheletro carbossilico del Nandrolone
Meccanismi putativi che migliorano i sintomi del dolore articolare:
Aumento della sintesi e del deposito di collagene nei tendini e nei legamenti.
Ritenzione del liquido sinoviale articolare.
Entrambi i meccanismi (aumento della produzione di collagene, ad esempio nei tendini flessori) e la ritenzione di liquido articolare sinoviale possono essere influenzati positivamente dal sistema renina-angiotensina (RAS). Il RAS regola l’equilibrio idrico ed elettrolitico, la crescita delle cellule del tessuto connettivo e il metabolismo del tessuto connettivo lasso e denso e dei siti di riparazione dei tessuti. [13]. Dal punto di vista patologico, l’attivazione del RAS aumenta la vascocostrizione, l’ipertrofia cardiaca e la fibrosi (con conseguente infarto del miocardio e fibrosi del fegato). [13]. Pertanto, è importante considerare la dualità del potenziale aumento dell’articolazione (cioè del tendine estensore del ginocchio) con l’uso di nandrolone: si può avere un beneficio transitorio nel rimodellamento del tendine (cioè del tendine estensore), ma attraverso questo stesso meccanismo, si possono accumulare disadattamenti fibrotici o cardiaci.
Quindi, per quanto riguarda i potenziali benefici transitori: L’enzima di conversione dell’angiotensina I (ACE) è un marcatore positivamente correlato all’attività dell’mRNA del collagene di tipo I e può riflettere il rimodellamento della matrice extracellulare (ECM) in cui la sintesi di collagene supera la degradazione.
Il Nandrolone aumenta l’attività ACE e incrementa la deposizione di collagene di tipo I nella matrice. In un modello di allenamento (ad esempio, pliometrico), Nandrolone + allenamento per i salti >> allenamento per i salti > sedentario per quanto riguarda l’attività ACE nel tendine (ad esempio, estensore del bicipite femorale), suggerendo una potenziale sinergia tra allenamento e Nandrolone a questo proposito. Si consideri, tuttavia, che questa stessa via è implicata nel rimodellamento del tessuto cardiaco e nell’azione patologica. Inoltre, l’aumento della rigidità tendinea rappresenta un rischio se non si aumenta la forza per ridurre la probabilità di lesioni gravi dovute a uno sforzo concentrato. Pertanto, le considerazioni sull’allenamento devono essere pianificate in modo rigoroso, soprattutto se si pratica l’allenamento pliometrico. [13].
Oxandrolone
Scheletro carbossilico del Oxandrolone
L’aumento significativamente maggiore della velocità di crescita in altezza ottenuto con il trattamento con GH più oxandrolone rispetto al solo GH si è riflesso in differenze simili nella risposta precoce dell’ALP ossea e del PICP, entrambi associati alla formazione dell’osso, ma non nel PIIINP, un marcatore del turnover dei tessuti molli, o nell’ICTP, un marcatore della degradazione del collagene osseo. Ciò suggerisce che l’oxandrolone può, direttamente o indirettamente, influenzare la proliferazione degli osteoblasti e la proliferazione e maturazione dei condrociti, con un effetto additivo rispetto a quello del solo GH. [2].
La reputazione del nandrolone di migliorare la funzione articolare durante le fasi di allenamento con carichi pesanti è rafforzata da questi risultati, secondo cui, aumentando l’attività dell’ACE e influenzando il RAS, serve ad aumentare la sintesi netta di collagene e l’equilibrio dei fluidi nelle articolazioni.
I risultati di Crofton et al. suggeriscono che GH+oxandrolone > GH+test > GH > placebo nel ΔPIINP, ma le differenze significative tra i gruppi potrebbero non essere misurabili a causa delle ridotte dimensioni del campione (un potenziale errore di tipo 2). [2].
La somministrazione di oxandrolone fino a 24 mesi a pazienti pediatrici gravemente ustionati ha migliorato significativamente il contenuto minerale osseo dell’intero corpo (WB BMC), il contenuto minerale osseo della colonna lombare (LS BMC), la densità minerale ossea della colonna lombare (LS BMD) e la velocità in altezza. [14].
Una grave ustione induce una risposta ipermetabolica e ipercatabolica caratterizzata da un aumento del lavoro cardiaco, del dispendio energetico a riposo e della degradazione delle proteine muscolari (1-6). Questa risposta compensatoria è accompagnata da un’elevata produzione epatica di glucosio e da insulino-resistenza (4, 5, 7-10). I pazienti in genere subiscono una perdita di massa magra e nei bambini la crescita è ostacolata. Nel tempo si verifica una significativa riduzione del contenuto minerale osseo (BMC), della densità minerale ossea (BMD) e del tessuto adiposo. [14].
Gli effetti sulla BMC/BMD sono diventati significativi in questi pazienti solo dopo più di un anno di trattamento continuo. [14]. Nei pazienti pediatrici è stata riscontrata una sinergia in questo effetto modulato dalla fase di maturazione della crescita, che potrebbe avere ramificazioni per l’uso in età adulta di rhGH e androgeni aromatizzanti, associati all’impennata puberale. L’ipotesi che si potrebbe trarre è che l’uso a lungo termine di androgeni aromatizzanti (ad esempio, T, nandrolone) in combinazione con dosi elevate di rhGH possa imitare alcuni aspetti della fase di maturazione della crescita nei bambini, potenzialmente aumentando la BMC e la BMD in modo sinergico in combinazione con l’oxandrolone se usato per lunghi periodi.
I particolari effetti dell’oxandrolone sul contenuto e sulla densità minerale ossea nei bambini in crescita suggeriscono che sia particolarmente utile nella fase iniziale (cioè i primi sei mesi) dell’uso continuo di RhGH, quando il turnover osseo è aumentato.
Stanozololo
Scheletro carbossilico dello Stanozololo
Lo Stanozololo, popolarmente associato al dolore articolare (“articolazioni doloranti e secche”), agisce sui fibroblasti sinoviali, precursori delle cellule che compongono le articolazioni sinoviali (ad esempio, anca, ginocchia, spalle), inibendo la sintesi del DNA. [10]. Mentre lo Stanozololo è considerato particolarmente potente nello stimolare l’attività della procollagenasi nel cuoio capelluto, un effetto che è associato all’aumento della secrezione di TGF-β1 [15] – stimolando così l’attività procollagene, ma inducendo perversamente, con questo stesso meccanismo, l’alopecia androgenica nel cuoio capelluto – l’effetto sulla cellula sinoviale è qualitativamente diverso.
L’inibizione della sintesi del DNA da parte dello Stanozololo nella cellula sinoviale, insieme ai suoi effetti sul C1-INH, fornisce diverse modalità esplicative per il fatto che provoca dolore nelle articolazioni sinoviali.
Alla fine del 1980 ricercatori britannici hanno scoperto che le cellule della pelle producono più collagene quando viene usato lo Stanozololo, ma che le cellule delle articolazioni non lo fanno [https://www.ncbi.nlm.nih.gov/pubmed/2556901].
Testosterone
Scheletro carbossilico del Testosterone
Il testosterone (250 mg a settimana) ha causato modesti aumenti dei biomarcatori del metabolismo del collagene, aumentando il PINP del 28%, l’ICTP del 22% e il PIINP del 70%. [16]. Il testosterone ha aumentato il tipo I (osso; PINP, ICTP) e il tipo III (tendini e legamenti, tessuti molli; PIINP) e ha potenziato in particolare l’effetto dell’rhGH sui marcatori di tipo III (PIINP), suggerendo un forte effetto modulante del testosterone sul collagene di tipo III che comprende tendini, legamenti e probabilmente fasce in risposta al GH. [16].
rhGH
Struttura peptidica del rhGH.
La somministrazione di rhGH nel tendine o nel legamento determina un marcato aumento del metabolismo del collagene con conseguente deposito netto. [17]. L’RhGH aumenta l’attività dell’osteocalcina e del procollagene di tipo I nel siero, riflettendo un aumento della BMD/BMC. Nel muscolo scheletrico e nel tendine la matrice extracellulare (ECM) conferisce importanti proprietà di trazione ed è di fondamentale importanza per la rigenerazione dei tessuti dopo una lesione. [10]. La somministrazione di rhGH promuove la sintesi di collagene ECM nel tessuto muscolo-tendineo di giovani adulti sani.
L’RhGH ha aumentato l’mRNA del collagene I (osso) di 2,3 volte e del collagene III (ECM, tendini, legamenti) di 2,5 volte. [10]. È stata osservata una tendenza ad un aumento di 5,8 volte della sintesi proteica del collagene muscolare. [10]. Il dosaggio utilizzato è stato di 33,3 µg * kg-¹ * giorno-¹ nei primi sette giorni e di 50 µg * kg-¹ * giorno-¹ in giovani uomini sani. [10]. Ciò equivale a una dose giornaliera di 3 – 4,5 UI di rhGH per un uomo di 90 kg.
Nella popolazione GHD si osserva una variazione bifasica (a due punte) della BMD in risposta alla somministrazione di rhGH, con una diminuzione iniziale a circa sei mesi dall’inizio della terapia, seguita da un successivo aumento dopo almeno un anno di trattamento. [18]. L’ipotesi prevalente è che il GH stimoli sia la formazione che il riassorbimento osseo, con conseguente aumento del turnover osseo. [18]. Questo effetto è evidente almeno nei primi sei mesi di somministrazione di rhGH, con conseguente diminuzione della BMD e del contenuto minerale osseo. [18]. Non è stata stabilita una relazione dose-risposta a causa dell’ampia variazione dei dosaggi utilizzati. [18]. Sembra che l’osso trabecolare (colonna vertebrale lombare) abbia una diversa sensibilità al GH rispetto all’osso corticale (collo del femore). [18].
L’RhGH si combina almeno in alcuni aspetti con gli androgeni (ad esempio, oxandrolone, testosterone) e in altri in modo sinergico per quanto riguarda il metabolismo dei tessuti molli. L’RhGH e il testosterone, se usati in combinazione, stimolano particolarmente la PIINP, suggerendo un effetto sinergico su tendini, legamenti e probabilmente fasce.
Conclusioni
Gli AAS hanno effetti di classe e specifici sulle articolazioni e sui tessuti connettivi che le compongono. In generale, gli AAS migliorano il metabolismo dei tessuti molli senza alcun effetto sul riassorbimento osseo, ma è importante considerare i vari effetti positivi e negativi degli AAS nei confronti dei tendini.
Gli androgeni aromatizzanti, come il testosterone, hanno effetti sinergici (più che additivi) in combinazione con il GH su alcuni aspetti del metabolismo del collagene, in particolare per quanto riguarda gli aspetti legati al metabolismo dei tessuti molli (ad esempio, la fascia), ed effetti additivi per altri aspetti.
I 17AA non aromatizzabili Oxandrolone (un 5α-androstan-3-one) e Stanozololo (un AAS il cui anello A ha una giunzione anulare pirazolica) condividono alcuni effetti di classe, ad esempio la stimolazione della produzione intrinseca di C1-INH che è associata a condizioni reumatologiche e artritiche, ma lo Stanozololo si distingue per la sua dimostrabile inibizione dose-dipendente della sintesi di DNA nelle cellule sinoviali che comprendono le principali articolazioni del corpo, modulando la disgregazione del tessuto connettivo. È stato dimostrato che l’Oxandrolone aumenta in modo additivo la densità minerale ossea (BMD/BMC) e può essere particolarmente utile nei primi sei mesi di inizio di un ciclo di androgeni aromatizzanti e rhGH per migliorare la conservazione della massa ossea.
Punto chiave: Un approccio sfumato al processo decisionale sul carico dell’allenamento (ad esempio, progressione, intensità, modalità) e sugli effetti dei farmaci (ad esempio, AAS, rhGH) in relazione alle articolazioni, ai tendini, ai legamenti e alle ossa, è reso possibile da una lettura attenta di questo articolo. Per i professionisti, le aree particolari da leggere con attenzione riguardano la rigidità dei tendini e le implicazioni per coloro che si impegnano in allenamenti pliometrici o anche con carichi leggeri; i pro e i contro degli AAS sugli aspetti biologici, strutturali e meccanici dei tendini; e la scarsità di dati solidi relativi agli effetti sui tessuti molli da parte di diversi AAS.
[1] Pärssinen, M., Karila, Kovanen, and Seppälä. (2000). The Effect of Supraphysiological Doses of Anabolic Androgenic Steroids on Collagen Metabolism. International Journal of Sports Medicine, 21(6), 406–411. doi:10.1055/s-2000-3834
[2] Crofton, P. M., Stirling, H. F., Schönau, E., and Kelnar, C. J. H. (1996). Bone alkaline phosphatase and collagen markers as early predictors of height velocity response to growth-promoting treatments in short normal children. Clinical Endocrinology, 44(4), 385–394. doi:10.1046/j.1365-2265.1996.706cn527.x
[3] Açil, Y., Brinckmann, J., Notbohm, H., Müller, P. K., and Bätge, B. (1996). Changes with age in the urinary excretion of hydroxylysylpyridinoline (HP) and lysylpyridinoline (LP). Scandinavian Journal of Clinical and Laboratory Investigation, 56(3), 275–283. doi:10.3109/00365519609088617
[4] Aerssens, J., Van Audekercke, R., Geusens, P., Schot, L. P. C., Osman, A. A.-H., and Dequeker, J. (1993). Mechanical properties, bone mineral content, and bone composition (collagen, osteocalcin, IGF-I) of the rat femur: Influence of ovariectomy and nandrolone decanoate (anabolic steroid) treatment. Calcified Tissue International, 53(4), 269–277. doi:10.1007/bf01320913
[5] Chidi-Ogbolu N, Baar K. Effect of Estrogen on Musculoskeletal Performance and Injury Risk. Front Physiol. 2019;9:1834. Published 2019 Jan 15. doi:10.3389/fphys.2018.01834
[6] Kjaer, M.J. (2008). Role of extracellular matrix in adaptation of tendon and skeletal muscle to mechanical loading. Physiol. Rev. 84:649-698.
[7] Joblecki, C.K., Heuser, J.E., Kaufman, S. (1973). Autoradiographic localization of new RNA synthesis in hypertrophying skeletal muscles. Journal of Cellular Biology. 57:743-759.
[8] Spadari, A., Romagnoli, N., Predieri, P. G., Borghetti, P., Cantoni, A. M., and Corradi, A. (2013). Effects of intraarticular treatment with stanozolol on synovial membrane and cartilage in an ovine model of osteoarthritis. Research in Veterinary Science, 94(3), 379–387. doi:10.1016/j.rvsc.2012.11.020
[9] Jones, I. A., Togashi, R., Rick Hatch, G. F., Weber, A. E., and Vangsness JR, C. T. (2018). Anabolic Steroids and Tendons: A Review of their Mechanical, Structural and Biologic Effects. Journal of Orthopaedic Research®. doi:10.1002/jor.24116
[10] Ellis AJ, Cawston TE, Mackie EJ. The differential effects of stanozolol on human skin and synovial fibroblasts in vitro: DNA synthesis and receptor binding. Agents Actions. 1994 Mar;41(1-2):37-43. doi: 10.1007/BF01986391. PMID: 8079819.
[11] Wright JK, Smith AJ, Cawston TE, Hazleman BL. The effect of the anabolic steroid, stanozolol, on the production of procollagenase by human synovial and skin fibroblasts in vitro. Agents Actions. 1989 Nov;28(3-4):279-82. doi: 10.1007/BF01967415. PMID: 2556901.
[12] Oleesky DA, Daniels RH, Williams BD, Amos N, Mor gan BP. Terminal complement complexes and C1/C1 inhibitor complexes in rheumatoid arthritis and other arthritic conditions. Clin Exp Immunol. 1991;84(2):250-255.
[13] Marqueti, R. de C., Hashimoto, N. Y., Durigan, J. L. Q., Batista e Silva, L. L., Almeida, J. A. de, Silva, M. da G. da, … Araújo, H. S. S. de. (2015). Nandrolone increases angiotensin-I converting enzyme activity in rats tendons. Revista Brasileira de Medicina Do Esporte, 21(3), 173–177. doi:10.1590/1517-869220152103143667
[14] Reeves, P. T., Herndon, D. N., Tanksley, J.D., Jennings, K., Klein, G. L., Mlcak, R. P., … Finnerty, C. C. (2016). FIVE-YEAR OUTCOMES AFTER LONG-TERM OXANDROLONE ADMINISTRATION IN SEVERELY BURNED CHILDREN. Shock, 45(4), 367–374. doi:10.1097/shk.0000000000000517
[15] Falanga, V., Greenberg, A. S., Zhou, L., Ochoa, S. M., Roberts, A. B., Falabella, A., and Yamaguchi, Y. (1998). Stimulation of Collagen Synthesis by the Anabolic Steroid Stanozolol. Journal of Investigative Dermatology, 111(6), 1193–1197. doi:10.1046/j.1523-1747.1998.00431.x
[16] Nelson, A. E., Meinhardt, U., Hansen, J. L., Walker, I. H., Stone, G., Howe, C. J., … Ho, K. K. (2008). Pharmacodynamics of Growth Hormone Abuse Biomarkers and the Influence of Gender and Testosterone: A Randomized Double-Blind Placebo-Controlled Study in Young Recreational Athletes. The Journal of Clinical Endocrinology and Metabolism, 93(6), 2213–2222.
[17] Vestergaard, P., Jørgensen, J. O. L., Olesen, J. L., Bosnjak, E., Holm, L., Frystyk, J., … Hansen, M. (2012). Local administration of growth hormone stimulates tendon collagen synthesis in elderly men. Journal of Applied Physiology, 113(9), 1432–1438. doi:10.1152/japplphysiol.00816.2012
[18] Barake, M., Klibanski, A., and Tritos, N. A. (2014). Effects of Recombinant Human Growth Hormone Therapy on Bone Mineral Density in Adults With Growth Hormone Deficiency: A Meta-Analysis. The Journal of Clinical Endocrinology and Metabolism, 99(3), 852–860. doi:10.1210/jc.2013-3921
Nelle precedenti parti di questo lungo viaggio alla scoperta e comprensione degli aminoacidi [Parte 1°, 2°, 3°, 4° e 5°] abbiamo capito chiaramente cosa sono, quali sono le loro funzioni biologiche ed abbiamo analizzato quegli AA che sono maggiormente utilizzati in campo sportivo. In questa parte conclusiva il viaggio raggiungerà il culmine con una trattazione approfondita degli Aminoacidi Essenziali/EAA.
Introduzione agli EAA:
Gli aminoacidi “essenziali” (EAA) della dieta – istidina, isoleucina, leucina, lisina, metionina, fenilalanina, treonina, triptofano e valina – sono detti “essenziali” perché non possono essere prodotti endogenamente e quindi devono essere consumati per la sopravvivenza umana. Inoltre, l’arginina è considerata un aminoacido “condizionatamente” essenziale, il che significa che in alcune circostanze la produzione endogena di arginina non riesce a soddisfare le esigenze fisiologiche. La necessità di consumare tutti gli EAA è stata ben stabilita negli ultimi 100 anni [1], ed esiste un fabbisogno giornaliero accettato per ciascun EAA come parte del normale apporto dietetico [2]. Il fabbisogno giornaliero si basa sulla quantità minima di ciascun EAA che deve essere consumata per evitare sintomi clinici di carenza. Un consumo inadeguato di uno solo dei nove EAA provoca sintomi di carenza, tra cui un’alterata sintesi proteica [3]. I requisiti per il consumo giornaliero di ciascun EAA sono convenzionalmente soddisfatti come componenti dell’apporto proteico dietetico di routine. La quantità e il profilo degli EAA nelle singole proteine alimentari, insieme alla digeribilità degli EAA legati alle proteine, costituiscono la base per la valutazione quantitativa della qualità della proteina pura [4]. Le proteine che contengono una quantità abbondante di tutti gli EAA in un formato altamente digeribile sono considerate proteine di “alta qualità” [4].
Sono stati fatti vari tentativi per esprimere la “qualità” o il “valore” di vari tipi di proteine. Le misure includono il valore biologico, l’utilizzo netto delle proteine, il rapporto di efficienza proteica, il punteggio aminoacidico corretto per la digeribilità delle proteine e il concetto di proteine complete. Questi concetti sono importanti nell’industria zootecnica, perché la mancanza relativa di uno o più aminoacidi essenziali negli alimenti per animali avrebbe un effetto limitante sulla crescita e quindi sul rapporto di conversione degli alimenti. Pertanto, diversi mangimi possono essere somministrati in combinazione per aumentare l’utilizzo netto delle proteine, oppure si può aggiungere al mangime un supplemento di un singolo aminoacido (metionina, lisina, treonina o triptofano).
Mentre l’importanza di soddisfare il fabbisogno minimo di ciascun EAA attraverso il consumo di proteine alimentari di alta qualità è riconosciuta da molti decenni [5], i benefici ottenibili dal consumo di EAA in forma libera in quantità superiori al fabbisogno minimo sono stati pienamente apprezzati solo negli ultimi 25 anni. Sono disponibili prodotti a base di singoli aminoacidi liberi, come la leucina o la lisina, e composizioni di piccoli gruppi di EAA, in particolare gli aminoacidi a catena ramificata (leucina, valina e isoleucina; BCAA), ma molti studi hanno documentato che si ottengono maggiori benefici da composizioni contenenti tutti gli EAA. L’integrazione giornaliera con composizioni di tutti gli EAA in forma libera ha dimostrato di essere benefica in molti modi [6]. In particolare, le composizioni di EAA in forma libera stimolano la sintesi proteica e il ricambio proteico nell’organismo, compresa la sintesi di nuove proteine muscolari. La stimolazione della sintesi proteica muscolare (MPS) da parte degli EAA può produrre un aumento della massa e della qualità muscolare, che si traduce in un miglioramento delle prestazioni fisiche e dei risultati funzionali [7].
La valutazione dei benefici degli EAA si differenzia da quella di molti altri integratori alimentari valutati in quanto esistono requisiti ben accettati per il consumo giornaliero di EAA. Inoltre, piuttosto che integratori contenenti un solo composto, come la creatina, esistono combinazioni quasi illimitate dei nove EAA che possono essere realizzate a seconda della richiesta fisiologica.
Meccanismo d’azione degli EAA
L’importanza del turnover delle proteine muscolari
Il continuo rinnovamento delle proteine muscolari degradate e danneggiate è importante per mantenere la massa e la funzione proteica del muscolo. Nello stato post-assorbitivo, la ripartizione netta delle proteine muscolari mantiene una fornitura costante di EAA plasmatici che forniscono precursori per la sintesi proteica in altri tessuti e organi. Gli EAA assunti con la dieta ripristinano la perdita netta di proteine muscolari stimolando la MPS. In condizioni normali, i tassi di MPS e di degradazione delle proteine muscolari sono uguali nel corso della giornata. Se la MPS supera il tasso di degradazione delle proteine muscolari, la massa muscolare aumenterà nel tempo, con un potenziale aumento della forza. L’accelerazione del turnover proteico muscolare (cioè la sintesi e la degradazione delle proteine aumentano in egual misura), senza un aumento della massa proteica muscolare netta, può anche giovare alla funzione muscolare, sostituendo le fibre muscolari più vecchie e danneggiate con nuove fibre altamente funzionanti [8]. Pertanto, la stimolazione del MPS/turnover è la principale base metabolica per l’aumento della forza e della funzione fisica. Sebbene anche i cambiamenti nella degradazione delle proteine giochino un ruolo nel controllo del metabolismo proteico muscolare, la stimolazione della MPS è la base principale degli effetti benefici degli integratori di EAA. Inoltre, le prime ricerche hanno dimostrato che l’effetto degli aminoacidi sul muscolo scheletrico si esplica principalmente attraverso la stimolazione della MPS, dato che la ripartizione delle proteine muscolari è rimasta invariata in uno studio acuto [9]. È importante notare che, poiché la misurazione della disgregazione delle proteine muscolari non è semplice e problematica in caso di assunzione esogena, la MPS rappresenta anche un indicatore surrogato del turnover proteico.
Controllo della sintesi proteica muscolare
Il modo più definitivo per valutare l’effetto degli integratori di EAA sulle prestazioni fisiche è quello di misurare le risposte metaboliche e funzionali nel tempo quando vengono fornite quantità e profili diversi di EAA, a condizione che tutte le altre variabili siano mantenute costanti. Tuttavia, per ottenere risultati affidabili possono essere necessari mesi di trattamento, a causa del lento tasso di turnover delle proteine muscolari e della difficoltà di controllare tutte le altre variabili (dieta, assunzione totale di EAA, attività, ecc.). Di conseguenza, l’uso della metodologia dei traccianti isotopici stabili per quantificare la risposta acuta delle MPS a una singola dose di EAA in soggetti umani è diventato il surrogato accettato per prevedere la risposta anabolica nel muscolo. Aspetti della sintesi proteica, trascrizione e traduzione, possono essere potenzialmente influenzati dal consumo di EAA: in particolare l’iniziazione e l’allungamento traslazionale (vedi Figura 1a). La trascrizione dell’RNA messaggero (mRNA) dal DNA comporta l’attivazione dei relativi geni. I cambiamenti nell’attivazione dei geni si riflettono nel numero di mRNA specifici nella cellula. L’espressione dell’mRNA è importante perché l’assemblaggio fisico di nuove proteine avviene sull’mRNA. Il complesso processo di iniziazione consiste in diverse fasi collegate tra loro e mediate da fattori di iniziazione eucariotici (eIF). Il complesso mammalian target of rapamycin 1 (mTORC1) è un regolatore chiave dell’attivazione degli eIF a valle che sono mediatori dell’iniziazione delle MPS (vedi Figura 1b). Sia la trascrizione che la traslazione del processo di sintesi proteica possono essere stimolate dagli aminoacidi e dall’esercizio fisico [13-16]. Tuttavia, sia la trascrizione dell’mRNA [17] che lo stato di fosforilazione di mTORC1 [18] sono generalmente poco correlati con i tassi di MPS, il che significa che nessuno dei due processi è probabilmente limitante per la MPS nella maggior parte delle circostanze. Il controllo traslazionale della sintesi proteica da parte della disponibilità di EAA è stato riconosciuto fin dal 1958 [19]. La traduzione comporta il collegamento successivo degli aminoacidi nell’ordine dettato dal codice dell’mRNA. Gli aminoacidi intracellulari liberi sono legati ai corrispondenti RNA di trasferimento (tRNA), formando molecole di tRNA cariche. Le molecole di tRNA cariche a loro volta trasferiscono in sequenza gli amminoacidi legati ai siti dell’mRNA che corrispondono al codice del tRNA carico. L’allungamento traslazionale può procedere fino al completamento solo se sono disponibili quantità adeguate di tutti i precursori amminoacidici richiesti. Una carenza relativa di un qualsiasi EAA lo renderà limitante e l’allungamento traslazionale verrà interrotto prima del completamento del processo. Il controllo traslazionale della MPS richiede che siano disponibili quantità adeguate di tutti gli EAA per sostenere un aumento dei tassi di MPS. Oltre a fornire i precursori necessari per la sintesi proteica, gli EAA aumentano i geni associati al rilevamento, al trasporto e alla regolazione di mTORC1 degli aminoacidi [20].
Segnalazione mTOR: regolazione di mTORC1 da parte di stimoli a monte; insulina, esercizio fisico (resistenza, endurance), glucosio e aminoacidi (Aa). L’esercizio fisico porta a un deficit energetico (aumento dell’AMP) che stimola l’AMPK, inibendo l’mTORC1, mentre il consumo di glucosio aumenta l’ATP, inibendo l’AMPK. L’insulina e l’esercizio di resistenza attivano la via PI3K, regolando positivamente mTORC1, mentre l’esercizio di resistenza attiva CaMK, promuovendo soprattutto la biogenesi mitocondriale. Gli AA stimolano principalmente mTORC1 promuovendo la fosforilazione e la de-fosforilazione delle GTPasi rag, rag A/B e rag C/D, rispettivamente. Gli AA stimolano generalmente FNIP1/FLCN, promuovendo la de-fosforilazione di rag C/D, tuttavia, alcuni EAA (leucina, istidina, valina, treonina, isoleucina, metionina) agiscono per promuovere la fosforilazione di rag A/B; ciò porta a un’upregulation di mTORC1 e della traduzione a valle, nonché del metabolismo del glucosio e dei lipidi. Le figure sono tratte da [10-12]. Abbreviazioni: Akt, proteina chinasi B; AMPK, proteina chinasi attivata dall’AMP; PI3K, fosfoinositide 3-chinasi; Ca2 +, ione calcio; CaMK, proteina chinasi calcio/calmodulina-dipendente; FNIP1, proteina folliculina-interagente 1; FLCN, folliculina (FLCN); mTORC1, complesso 1 del bersaglio mammifero della rapamicina; FOX-O, fattori di trascrizione forkhead box-O; PGC-1α, peroxisome proliferator-activated gamma coactivator-1 alpha; MuRF-1, muscle ring-finger protein-1; eEF-2K, eukaryotic elongation factor-2 kinase; eEF2, eukaryotic elongation factor-2; TSC1/2, Tuberous sclerosis proteins 1 (hamartin) +2 (tuberin); Rheb, Ras homolog enriched in brain; LRS, leucil-tRNA sintetasi; SESN2, Sestrin-2; GATOR1/2, GAP (GTPase-activating protein) activity toward Rags 1+2; SAM, s-adenosyl methionine; SAMTOR, s-adenosyl methionine sensor for mTORC1; 4E-BP1, eukaryotic translation initiation factor 4E-binding protein 1; P70-S6K, (S6K1) proteina ribosomiale S6 chinasi beta-1; SREBP, sterol regulatory element binding protein; HIF-1α, hypoxia-inducible factor-1 alpha; PDCD4, Programmed cell death protein 4; SKAR, S6K1 Aly/REF-like substrate; eIF4E/B, eukaryotic translation initiation factor 4E+B.Sintesi proteica: iniziazione, allungamento e terminazione trascrizionale che portano alla produzione di mRNA nel nucleo, poi esportato nel citosol per essere sottoposto a iniziazione, allungamento e terminazione traslazionale; produce un polipeptide che viene ripiegato in una proteina.
Importanza del protocollo utilizzato per misurare la risposta delle MPS al consumo di aminoacidi
Il metodo più comunemente utilizzato per misurare la MPS nei soggetti umani consiste nel determinare il tasso di incorporazione di un aminoacido tracciante con isotopi stabili nelle proteine muscolari nel corso del tempo, diviso per l’arricchimento del precursore [21]. Questo approccio porta al calcolo del tasso di sintesi frazionale delle proteine muscolari (FSR). Poiché la massa muscolare è relativamente costante nell’arco di diversi giorni, le variazioni del FSR sono convenzionalmente considerate un riflesso diretto della MPS [21]. Un approccio alternativo alla misurazione della MPS si basa sulla differenza artero-venosa di traccianti aminoacidici non marcati e marcati e sull’arricchimento isotopico del pool libero intracellulare [22]. Questi due metodi danno risultati comparabili per la MPS in soggetti umani [23]. Infatti, è stato dimostrato che la stimolazione acuta della MPS da parte del consumo di EAA si riflette nel bilancio proteico delle 24 ore della gamba [24]. L’evidenza supporta la traduzione delle differenze nella risposta acuta della MPS al consumo di EAA in parametri di risultato misurati per settimane o mesi. Ad esempio, l’effetto del consumo giornaliero di una formula a base di EAA in soggetti sani giovani e normali è stato confrontato con un placebo per 28 giorni di riposo completo a letto [7]. Il consumo quotidiano della formula a base di EAA per tutti i 28 giorni di riposo a letto ha migliorato la perdita di massa muscolare osservata nei soggetti che consumavano il placebo di una quantità prevista dallo studio con tracciante prima del riposo a letto [7]. L’accuratezza predittiva del metodo del tracciante in acuto in questo paradigma è particolarmente significativa perché l’attività e l’assunzione di cibo sono state completamente controllate durante i 28 giorni di intervento [7].
Il periodo di tempo in cui viene determinata la MPS è importante per interpretare il significato fisiologico dei cambiamenti acuti in risposta all’assunzione di aminoacidi. Un aumento transitorio della MPS ha meno probabilità di predire un aumento a lungo termine della massa e della funzione muscolare rispetto a una risposta che rimane al di sopra del valore di base per tre ore o più. Ad esempio, il consumo di leucina da sola può suscitare una risposta transitoria (1-2 ore) nella MPS muscolare, ma questa risposta deve essere interpretata con cautela [25]. Il consumo di una quantità sufficiente di leucina da sola può attivare mTORC1 e le molecole associate coinvolte nell’avvio del processo di sintesi proteica e si riflette in un aumento transitorio delle proteine muscolari MPS. Tuttavia, la sintesi delle proteine muscolari richiede un’adeguata disponibilità di tutti gli aminoacidi componenti, compresi i nove EAA. In assenza di apporto dietetico, gli EAA necessari per produrre proteine muscolari complete devono provenire da fonti endogene. Inizialmente, gli EAA aggiuntivi necessari per la sintesi di proteine muscolari complete possono provenire da pool di EAA liberi nel fluido intracellulare ed extracellulare. Tuttavia, il conseguente esaurimento degli EAA liberi in questi pool limiterà la sintesi proteica muscolare a causa dell’inadeguata disponibilità di precursori (EAA disponibili nei pool di aminoacidi). L’unica altra fonte potenziale degli EAA necessari per mantenere la MPS in questa circostanza è la degradazione proteica accelerata, che limiterà qualsiasi guadagno netto di proteine muscolari che ci si potrebbe aspettare in base alle variazioni acute della MPS. Pertanto, la risposta anabolica (cioè MPS – MPB) delle proteine muscolari al consumo di un singolo EAA, come la leucina, o di piccoli gruppi di EAA (BCAA) sarà limitata dalla scarsa disponibilità degli altri EAA.
Il lavoro di Fuchs e collaboratori [26] fornisce prove sull’interpretazione dell’importanza dell’intervallo di campionamento sulla risposta della FSR muscolare all’assunzione di aminoacidi. In questo studio, la FSR muscolare è stata determinata in risposta al consumo di BCAA, proteine del latte o chetoacidi a catena ramificata, corrispondenti a leucina, valina e isoleucina. La FSR delle proteine muscolari è stata stimolata nelle prime due ore dopo il consumo di tutti e tre gli integratori alimentari. Tuttavia, 2-5 ore dopo l’ingestione di ciascun integratore, la FSR delle proteine muscolari è rimasta stimolata solo dopo il consumo di proteine del latte. In altre parole, l’ingestione dei BCAA o dei chetoacidi associati non è riuscita a stimolare la FSR delle proteine muscolari rispetto al valore di base dopo il consumo [26]. Il tasso di FSR delle proteine muscolari è stato limitato nelle ore 2-5 dopo il loro consumo da una diminuzione della disponibilità degli EAA non forniti dall’integratore alimentare, come risulta dalla diminuzione della concentrazione plasmatica di fenilalanina. La diminuzione degli EAA plasmatici è stata probabilmente attenuata in qualche misura da un aumento del tasso di degradazione delle proteine muscolari, limitando così l’effetto anabolico netto della stimolazione della FSR delle proteine muscolari. Al contrario, la disponibilità di fenilalanina (come riflesso degli EAA) era elevata 2-5 ore dopo il consumo di proteine del latte a causa della continua digestione e assorbimento di tutti gli EAA, nonché della disponibilità di aminoacidi non essenziali di supporto. Gli autori hanno concluso che “questi dati suggeriscono che, oltre all’aumento postprandiale delle concentrazioni plasmatiche di BCAA, è necessario fornire altri aminoacidi (essenziali) per consentire un aumento postprandiale più prolungato del tasso di sintesi proteica muscolare” [26]. È quindi ragionevole basarsi principalmente su dati provenienti da studi in cui la MPS è stata determinata in un intervallo di 3 ore o più dopo il consumo di EAA per aspettarsi una traduzione dei risultati in risultati funzionali.
Sicurezza
Il consumo di EAA non è stato segnalato come causa di reazioni avverse. I soggetti affetti da rare malattie genetiche che comportano un’alterazione della capacità di metabolizzare alcuni EAA, come la malattia delle urine a sciroppo d’acero (incapacità di metabolizzare i BCAA), potrebbero avere una risposta avversa agli integratori contenenti tutti gli EAA. Tuttavia, gli errori innati del metabolismo che influenzano il metabolismo di un EAA sono evidenti in età precoce e l’adattamento alla dieta è necessario per la salute ed eventualmente per la sopravvivenza. È quindi improbabile che un adulto con un errore innato del metabolismo che limita il consumo sicuro di EAA non sia consapevole di tale condizione. È anche possibile che un individuo con una malattia renale possa reagire male all’integrazione di EAA, poiché una dieta a basso contenuto proteico è spesso raccomandata nelle malattie renali a causa dell’accumulo di urea e ammoniaca nel sangue. Tuttavia, un’integrazione a base di EAA generalmente non contribuisce ad aumentare la produzione di urea o ammoniaca a causa del maggiore riutilizzo di aminoacidi non essenziali per la sintesi proteica. Tuttavia, non sono disponibili dati sufficienti su individui con funzionalità renale compromessa per determinare la sicurezza degli integratori alimentari a base di EAA.
Sono disponibili pochi dati su cui basare il limite massimo di sicurezza del consumo dei singoli EAA. La Tabella 1 elenca i livelli di consumo di ciascun EAA che si sono dimostrati sicuri. I limiti massimi di sicurezza riportati nella Tabella 1 sono espressi come quantità di ciascun EAA consumata al di sopra dell’assunzione abituale. Pertanto, se si considerano le quantità sicure di ciascun EAA, questi dati indicano che più di 100 g di EAA supplementari possono essere consumati in modo sicuro al giorno in un adulto americano che già consuma l’apporto alimentare medio abituale di circa 40 grammi al giorno. Il dosaggio ragionevole di un integratore di EAA non supera i 15 g, il che significa che anche tre dosi massime al giorno sono in linea con il normale consumo giornaliero di EAA attraverso le fonti alimentari di proteine. I seguenti dati sugli effetti dell’integrazione di EAA sono stati ricavati in popolazioni con un’adeguata assunzione di proteine alimentari, a meno che non sia indicato diversamente.
Fabbisogno alimentare di aminoacidi essenziali e limite massimo di consumo sicuro per gli adulti:
Riferimenti [2,4], basati sui DRI; 2Al di sopra dell’assunzione abituale.
Punti chiave:
Il turnover proteico assicura il continuo rinnovo delle proteine muscolari degradate e danneggiate ed è importante per mantenere la massa e la funzione proteica muscolare.
Il surrogato accettato per la misurazione del turnover proteico è la determinazione della sintesi proteica muscolare con la metodologia dei traccianti isotopici stabili. Sebbene la degradazione delle proteine sia importante in questo processo, la risposta acuta principale dell’assunzione di EAA sul muscolo scheletrico è la stimolazione della sintesi proteica.
Il limite massimo di sicurezza dell’assunzione giornaliera di EAA consente un’integrazione sostanziale.
Consenso dei risultati della ricerca
EAA e sintesi proteica muscolare a riposo
La MPS è stimolata dal consumo di composizioni di EAA [35] e inibita da una ridotta disponibilità di EAA nel plasma [36]. L’entità dell’aumento della MPS in seguito al consumo di EAA è funzione della quantità ingerita. A riposo, è stato riportato che una dose orale di EAA pari a 1,5 g stimola la MPS [37], mentre la dose massima efficace, dopo la quale non si ottiene un’ulteriore stimolazione della sintesi in una singola dose, è ritenuta pari a 15-18 grammi di EAA [38]. La stimolazione delle MPS attraverso il consumo di EAA non richiede il consumo simultaneo di aminoacidi non essenziali (NEAA) [35,38]. L’inclusione di NEAA in una miscela di 18 g di EAA nel profilo di proteine di manzo non ha avuto alcun effetto sulla stimolazione mediata dagli EAA della MPS [35]. Mentre il consumo di NEAA non ha alcun effetto sulla MPS quando si consumano meno di 18 g di EAA, è possibile che quando si consumano più di 18 g di EAA i NEAA siano limitati dai tassi massimi di produzione endogena; tuttavia, sono necessarie ulteriori ricerche per esplorare questa possibilità.
Gli integratori di EAA stimolano le MPS più di una pari quantità di proteine di alta qualità, sia come isolato [39] sia come componente di un pasto [40]. È stato riscontrato che una dose orale di 3 g di EAA stimola le MPS in misura simile a 20 g di proteine isolate del siero di latte, che contengono circa 10 g di EAA [41]. Inoltre, è stato dimostrato che l’aggiunta di EAA alle proteine del siero di latte aumenta significativamente la risposta delle MPS rispetto alle sole proteine del siero di latte [42]. L’effetto stimolante superiore degli EAA in forma libera (solo singoli EAA) è legato alla maggiore quantità di EAA/grammo rispetto a una fonte proteica alimentare [43]. A causa dell’elevato tasso di assorbimento intestinale degli EAA in forma libera [44], il rapido aumento delle concentrazioni plasmatiche di EAA in circolo favorisce il trasporto verso l’interno del muscolo [23,45], con conseguente raggiungimento più rapido del picco di concentrazione intramuscolare di EAA rispetto ad altre fonti proteiche alimentari.
L’importanza delle concentrazioni plasmatiche di EAA e della velocità di aumento fino al picco di concentrazione sulla risposta MPS non è chiara, ma alcuni studi hanno trovato una relazione tra le concentrazioni plasmatiche di EAA e la MPS [46] e l’analisi di dati consolidati mostra una correlazione tra la velocità di aumento fino al picco di concentrazione di EAA e la MPS [43]. D’altra parte, non è stata riscontrata alcuna differenza nella risposta alla MPS quando la stessa dose di EAA è stata somministrata in un unico bolo o in cinque dosi più piccole nel tempo [47]. Pertanto, si concorda sull’esistenza di una relazione tra la dose di EAA e la risposta alle MPS, ma non si concorda sui meccanismi di regolazione che collegano dose e MPS.
L’impatto complessivo di un integratore alimentare sulla MPS nell’arco delle 24 ore dipende non solo dalla risposta acuta al consumo della composizione, ma anche dalle risposte anaboliche ai pasti normali. È ormai assodato che la risposta anabolica a un pasto è ridotta in seguito a un pre-carico con un isolato proteico [48]. Al contrario, una dose di 15 g di EAA non ha avuto alcun impatto sulla risposta anabolica al pasto successivo [49]. Tuttavia, è importante notare che l’integrazione di EAA in forma libera determina un aumento delle concentrazioni ematiche di EAA molto maggiore rispetto a un pasto con una quantità maggiore di EAA, poiché non richiedono digestione e vengono assorbiti rapidamente. La Figura 2 mostra gli effetti di 15 g di EAA in forma libera [50] sulle concentrazioni plasmatiche rispetto a un pasto misto contenente 70 grammi di proteine di manzo [51]. A causa della lenta digestione e del rilascio degli EAA alimentari dopo il consumo di un pasto misto, i livelli ematici aumentano solo in minima parte, rispetto a un aumento rapido e robusto dopo l’ingestione di EAA cristallini/liberi. Inoltre, questo rapido aumento (3 volte o più) predice una maggiore risposta della MPS [43].
L’effetto di 15 g di EAA in forma libera rispetto a 70 g di proteine magre di manzo e all’ingestione di un pasto misto sulla cinetica plasmatica degli EAA. Adattato dalle referenze [50,51].
Lo stato fisiologico può influenzare la risposta delle MPS agli EAA. L’invecchiamento è lo stato non clinico più comunemente studiato in cui la risposta agli EAA può essere alterata, definita resistenza anabolica. Una minore reattività delle MPS al consumo è stata ben documentata [52,53], ma non osservata in modo coerente [54]. Le diverse risposte delle MPS degli individui anziani al consumo di EAA possono essere spiegate da differenze nel profilo EAA della composizione. Ad esempio, in un’occasione Katsanos et al. hanno fornito a soggetti anziani una miscela di 6,7 g di EAA con il profilo presente nelle proteine del siero di latte (27% di leucina) e in una seconda occasione hanno fornito la stessa quantità di una miscela di EAA con la leucina che comprendeva circa il 40% del totale degli EAA [55]. In questo studio, la composizione al 40% di leucina ha stimolato la sintesi proteica muscolare circa il 50% in più rispetto al profilo di leucina inferiore, nonostante contenesse la stessa quantità di EAA totali [55]. Questi risultati dimostrano la potenziale importanza del profilo EAA in una composizione. Tuttavia, le combinazioni dei nove EAA sono pressoché illimitate e ci sono pochi dati che confrontano direttamente l’efficacia di diversi profili di EAA nella stessa circostanza. A causa della mancanza di dati comparativi sufficienti, l’impatto dei diversi profili di EAA sulla MPS non sarà discusso in questo documento. A questo proposito sono necessarie ulteriori ricerche per determinare i profili ottimali per i vari requisiti fisiologici.
EAA e bilancio proteico ed energetico dell’intero organismo
Un bilancio netto negativo delle proteine in tutto il corpo diverse da quelle muscolari (cioè nei tessuti e negli organi; riflesso dai tassi di sintesi e di degradazione delle proteine nell’intero organismo) influisce negativamente sulle proteine muscolari e quindi sulle prestazioni fisiche. Se l’assunzione di precursori di EAA con la dieta non è sufficiente a soddisfare il fabbisogno di tutto l’organismo, la scomposizione delle proteine muscolari e il rilascio di aminoacidi nel sangue forniranno gli EAA necessari. Un bilancio energetico negativo influirà indirettamente anche sulle proteine muscolari, poiché gli EAA ingeriti saranno almeno in parte destinati all’ossidazione per la produzione di energia, anziché essere incanalati verso la sintesi proteica muscolare [56]. Una discussione sugli effetti degli EAA sulla sintesi proteica muscolare deve quindi essere considerata nel contesto dello stato dell’equilibrio proteico ed energetico dell’intero organismo.
I periodi di deficit calorico sono comuni nelle categorie di peso e negli sport di resistenza come la maratona e il nuoto di distanza, dove i periodi di allenamento intenso e il desiderio di avere un peso corporeo ridotto possono limitare l’apporto calorico [57]. Il deficit calorico aumenta il fabbisogno di EAA dell’intero organismo [58]. Ad esempio, cinque giorni di deficit calorico del 30% hanno richiesto un aumento di 3 volte dell’assunzione di EAA per produrre un bilancio proteico corporeo positivo [58]. Se non si riesce a soddisfare l’aumento del fabbisogno di EAA dell’intero corpo, si verifica una scomposizione netta delle proteine muscolari per fornire gli EAA necessari e non si può invertire completamente la tendenza fino a quando non si soddisfa il fabbisogno dell’intero corpo.
Molti stati clinici inducono cambiamenti nel metabolismo proteico dell’intero corpo che influenzano il fabbisogno di EAA e l’equilibrio proteico muscolare. Possono esserci nuove richieste di precursori di EAA per funzioni quali la riparazione dei tessuti danneggiati, la guarigione delle ferite e la produzione di proteine della fase acuta. In queste condizioni è probabile che la normale risposta anabolica del muscolo scheletrico agli EAA assunti con la dieta diminuisca (resistenza anabolica). Di conseguenza, la rapida perdita di massa muscolare è una complicazione comune di gravi malattie e lesioni [59]. La stessa risposta può verificarsi, anche se in misura minore, in seguito a un allenamento intenso o a un evento agonistico, in particolare negli atleti che si trovano volontariamente o involontariamente in deficit calorico.
Punti chiave: Effetti degli EAA sul muscolo e sulle proteine dell’intero corpo
Esiste una dose-risposta degli EAA orali sulla sintesi proteica del muscolo scheletrico che raggiunge un plateau a circa 15-18 g.
Esiste una relazione tra la cinetica degli EAA plasmatici e la stimolazione della sintesi proteica.
Gli EAA orali stimolano la sintesi proteica muscolare in misura maggiore rispetto a una pari quantità di proteine di alta qualità.
La riduzione della risposta anabolica con l’invecchiamento richiede un diverso profilo di EAA, in particolare una maggiore proporzione di leucina.
Il fabbisogno di EAA dell’intero organismo aumenta con il deficit calorico. Se questo fabbisogno non è soddisfatto, si verifica una disgregazione netta delle proteine muscolari per fornire gli EAA necessari.
EAA e funzione fisica in assenza di esercizio fisico
Diversi studi documentano che la stimolazione acuta delle MPS da parte di composizioni libere di EAA si traduce in un aumento a lungo termine della massa e della funzione muscolare, anche in assenza di un controllo dell’assunzione di proteine con la dieta. Gli studi sui risultati sono stati generalmente condotti su individui anziani. Utilizzando un disegno randomizzato, in doppio cieco e controllato con placebo, donne anziane sono state assegnate a ricevere placebo o 15 g di EAA al giorno per tre mesi [60]. L’ingestione di 7,5 g di EAA ha stimolato acutamente la FSR delle proteine muscolari in entrambi i gruppi al basale [60]. La FSR basale a tre mesi era aumentata solo in coloro che ricevevano un’integrazione giornaliera di EAA, e l’entità della risposta acuta agli EAA era inalterata dopo tre mesi di consumo di EAA. Coerentemente con i dati sulla FSR delle proteine muscolari, la massa corporea magra è aumentata significativamente nei soggetti che ricevevano EAA ma non il placebo [36]. In uno studio simile, 12 soggetti intolleranti al glucosio hanno ingerito 11 g di EAA due volte al giorno tra i pasti per 16 settimane [61]. La dieta e l’attività fisica non sono state modificate in altro modo. Il consumo di EAA ha aumentato la massa magra e, soprattutto, ha migliorato una serie di parametri della funzione fisica [61]. In un gruppo di 38 donne anziane (≥75 anni), l’integrazione quotidiana con 3 g di EAA due volte al giorno per tre mesi ha migliorato significativamente la velocità di camminata [62]. Risultati simili sono stati osservati in uno studio che ha incluso 92 persone anziane a bassa funzionalità a cui sono stati somministrati per 12 settimane integratori di 15 g di proteine isolate del siero di latte, EAA (12 g di EAA più 3 g di aromi) o educazione alimentare [63]. I soggetti che hanno ricevuto gli EAA hanno migliorato significativamente la distanza percorsa in 6 minuti, la forza di presa e la forza delle gambe (coppia di picco misurata con il Cybex). Anche i soggetti che hanno ricevuto le proteine del siero del latte hanno migliorato significativamente la distanza percorsa in 6 minuti, ma il miglioramento è stato significativamente inferiore rispetto a quelli che hanno ricevuto gli EAA. La forza delle gambe non è migliorata nel gruppo del siero di latte. È interessante notare che la distanza percorsa dal gruppo di educazione alimentare è diminuita nel corso delle 12 settimane di intervento [63]. A riprova del potenziale impatto positivo dell’integrazione di EAA sui miglioramenti funzionali, l’entità del miglioramento osservato nella distanza di cammino di 6 minuti nei soggetti che ricevevano EAA era approssimativamente la stessa riportata in una revisione sistematica di studi che riportavano i risultati di 2-6 mesi di allenamento di resistenza in 241 individui [64]. Questi risultati sono coerenti con l’estrapolazione degli effetti acuti della somministrazione di EAA sul controllo della MPS [46] attraverso la farmacocinetica ematica [43]. In uno studio condotto su persone anziane e sane costrette al riposo a letto per 10 giorni, il consumo di tre dosi di 15 g di EAA al giorno ha attenuato il declino della funzione fisica evidente nei soggetti che avevano ricevuto un placebo equivalente dal punto di vista calorico [65]. In sintesi, gli studi esistenti sugli effetti degli EAA sui risultati funzionali in assenza di allenamento si sono concentrati in gran parte su popolazioni anziane o compromesse. Poiché gli EAA sono potenti stimolatori dell’anabolismo proteico muscolare e dell’intero corpo, queste popolazioni sono logiche destinatarie di indagini primarie in quanto manifestano una resistenza anabolica nel muscolo scheletrico che porta alla perdita muscolare, alla debolezza funzionale, alle comorbidità e ad altri esiti clinici negativi. Pertanto, è opportuno raccomandare ulteriori studi per determinare l’effetto anabolico degli EAA in giovani individui sani in assenza di allenamento.
Interazione degli EAA con l’esercizio fisico
I primi esami incrociati degli effetti degli aminoacidi sul muscolo scheletrico hanno dimostrato che l’aumento della somministrazione di aminoacidi a riposo determina una stimolazione del trasporto di aminoacidi verso l’interno del muscolo scheletrico (dal 30 al 100%, a seconda dei singoli EAA), una stimolazione della sintesi proteica (dal 30 al 300%) e un miglioramento del bilancio netto di aminoacidi [9]. Quando l’esercizio di resistenza della parte inferiore del corpo è stato eseguito prima dell’infusione di aminoacidi, si sono verificati effetti maggiori nel trasporto verso l’interno di alcuni aminoacidi, ma soprattutto si sono registrati aumenti ancora maggiori nella sintesi proteica e nel bilancio proteico muscolare netto [9]. In ogni caso, non si sono verificati cambiamenti nella degradazione delle proteine muscolari. È importante notare che gli effetti combinati dell’esercizio di resistenza e dell’aumento dell’apporto di aminoacidi sono interattivi. Quando lo stesso esercizio di resistenza è stato eseguito senza la somministrazione di aminoacidi, si è registrato un aumento del trasporto interno di aminoacidi e della sintesi proteica muscolare; tuttavia, il bilancio muscolare netto è rimasto negativo [66]. L’assenza di miglioramento del bilancio netto è dovuta all’aumento della sintesi e della degradazione delle proteine dopo il solo esercizio di resistenza [66]. Pertanto, l’esercizio di resistenza da solo non porta all’anabolismo muscolare (il bilancio netto delle proteine muscolari è negativo). L’anabolismo si verifica solo se supportato dai precursori amminoacidici necessari. La Figura 3 rappresenta graficamente il bilancio trasversale degli arti della fenilalanina (un surrogato del bilancio degli aminoacidi, poiché non viene metabolizzata nel muscolo scheletrico) a digiuno, dopo il solo esercizio di resistenza, dopo l’infusione dei soli aminoacidi e con l’infusione combinata di aminoacidi ed esercizio di resistenza. Gli effetti interattivi degli EAA somministrati per via orale e dell’esercizio di resistenza hanno rivelato risultati simili. L’esercizio di resistenza della parte inferiore del corpo è stato seguito dalla somministrazione di EAA per via orale o di una miscela completa di aminoacidi. I risultati hanno indicato che gli EAA dopo l’esercizio fisico hanno migliorato il bilancio netto muscolare nella stessa misura della miscela completa, fornendo ulteriori prove [38] del fatto che, se somministrati insieme all’esercizio fisico, solo gli EAA sono necessari per stimolare l’anabolismo muscolare [67]. Anche l’ingestione orale in bolo di EAA dopo l’esercizio di resistenza della parte inferiore del corpo ha aumentato la sintesi proteica e il bilancio netto muscolare, indipendentemente dal fatto che la bevanda sia stata consumata una o tre ore dopo l’esercizio [68]. Vi sono indicazioni che i benefici degli EAA sul muscolo scheletrico potrebbero non dipendere interamente dalla stimolazione delle MPS. L’integrazione di EAA ha migliorato l’evidenza istologica del danno muscolare e ha ridotto la perdita di forza muscolare, anche in assenza di cambiamenti nella MPS [69].
Bilancio netto muscolare di fenilalanina (umol/kg/min) durante il digiuno, l’esercizio di resistenza da solo (RE), la somministrazione/infusione completa di aminoacidi da sola (AA) e con la combinazione di AA e RE. È stato dimostrato un effetto interattivo tra la somministrazione di RE e AA. Dati derivati dalle referenze [9,66].
Gli effetti interattivi degli EAA e dell’esercizio di resistenza si riflettono nella segnalazione dell’avvio della traduzione nel muscolo. Ai volontari è stata somministrata una soluzione di placebo, leucina, BCAA o EAA dopo l’esercizio contro resistenza. I risultati hanno indicato che 90 minuti dopo il recupero dell’esercizio, l’attivazione della proteina ribosomiale S6K1 e del fattore di iniziazione della traduzione eucariotica 4E-BP1, nonché una riduzione sostenuta dell’interazione 4E-BP1:eIF4E, erano maggiori con gli EAA [70]. Mentre nello studio sugli EAA è stato osservato un aumento di 9 volte dell’espressione di S6K1, la stimolazione complessiva dell’iniziazione della traduzione è stata più efficace con gli EAA, con conseguente aumento progressivo dell’iniziazione della traduzione (placebo < leucina < BCAA < EAA) [70]. Uno studio condotto su giovani uomini ha confermato l’aumento della via di segnalazione mammalian target of rapamycin complex 1 (mTORC1) dopo l’esercizio di resistenza, ma ha anche osservato che un integratore di EAA mantiene mTORC1 nelle regioni periferiche delle fibre muscolari, più vicino al suo attivatore diretto Rheb [71]. Gli autori hanno ipotizzato che “la localizzazione intracellulare di mTOR può servire a innescare la chinasi per futuri stimoli anabolici” [71]. Gli effetti degli EAA e dell’esercizio di resistenza sull’aumento della segnalazione anabolica sono coerenti con l’invecchiamento. Gli EAA e l’esercizio di resistenza aumentano le concentrazioni periferiche di EAA in egual misura sia nei soggetti giovani che in quelli più anziani [72]. Indipendentemente dall’età, con il trattamento combinato sono stati dimostrati aumenti di mTOR (Ser2481) e della proteina ribosomiale S6 (Ser235/236) [72], il che suggerisce una maggiore sensibilità muscolare agli stimoli combinati. inoltre, la somministrazione di EAA dopo l’esercizio fisico in uomini anziani porta a un aumento della proliferazione delle cellule satelliti [73]. Il prodotto metabolico di questa segnalazione è un miglioramento della sintesi proteica muscolare. Inoltre, donne anziane hanno eseguito un’estensione unilaterale del ginocchio e poi hanno consumato 1,5 o 6 g di una formula EAA con quantità variabili di leucina, oppure 20 o 40 g di proteine del siero di latte. I risultati hanno indicato che le dosi di 1,5 g e 6 g di EAA erano efficaci quanto 40 g di proteine del siero del latte nella stimolazione della sintesi proteica muscolare acuta (miofibrillare) [37].
Strategie di integrazione: Tempistica degli EAA
È necessario considerare la tempistica della somministrazione di EAA in relazione all’esercizio di resistenza. I soggetti a cui sono stati somministrati EAA immediatamente prima o dopo l’esercizio di resistenza hanno entrambi registrato un aumento del 130% delle concentrazioni di fenilalanina arteriosa e muscolare [74]. Tuttavia, l’effetto sull’assorbimento netto di fenilalanina (una misura indiretta della sintesi proteica netta) è stato molto maggiore quando la bevanda è stata consumata subito prima dell’esercizio [74]. È importante notare che ogni trattamento ha portato a un bilancio netto di fenilalanina positivo; tuttavia, il migliore apporto di aminoacidi (flusso sanguigno X concentrazione arteriosa di EAA) quando è stato consumato immediatamente prima dell’esercizio ha portato a un apporto di aminoacidi circa 3 volte superiore [74]. L’effetto stimolante dell’esercizio di resistenza sul flusso sanguigno muscolare, se combinato con il maggiore apporto di aminoacidi derivante dal consumo prima dell’esercizio, determina una maggiore risposta anabolica nel muscolo scheletrico. Ciò è coerente con una recente revisione che denota la relazione tra l’aumento degli EAA periferici e la stimolazione della sintesi proteica muscolare e dell’intero corpo [43]. Altri lavori non confermano questi risultati, poiché una soluzione di carboidrati/EAA somministrata prima dell’esercizio di resistenza non ha migliorato la sintesi proteica muscolare post-esercizio [75]; tuttavia, le differenze metodologiche/interpretative e l’assunzione di carboidrati possono complicare la coerenza dell’interpretazione. Inoltre, è stato dimostrato che un secondo bolo di EAA un’ora dopo il primo duplica l’anabolismo muscolare della prima somministrazione, indicando che i meccanismi di sintesi non sono inattivi dopo una prima stimolazione [76].
Interazione degli EAA con altre modalità di esercizio
A causa dei forti effetti interattivi dimostrati con gli EAA e l’esercizio di resistenza, la maggior parte del lavoro si è concentrata su questa combinazione. Anche i dati sulla combinazione di EAA ed esercizio aerobico sono coerenti con i suoi effetti sull’anabolismo proteico del muscolo scheletrico. Nei giovani adulti che eseguivano 90 minuti di esercizio in cicloergometria o con carico ponderato (30% della massa corporea) su tapis roulant, con o senza EAA (consumati ogni 30 minuti per tutta la durata dell’esercizio), la sintesi proteica muscolare era maggiore durante ciascuna modalità di esercizio con gli EAA [77]. Tuttavia, il trasporto del carico e gli EAA hanno determinato un aumento maggiore della MPS sia durante che dopo l’esercizio [77]. Questi risultati indicano che il carico del muscolo scheletrico fornisce uno stimolo più forte per gli effetti combinati di EAA ed esercizio fisico. L’interazione tra EAA ed esercizio aerobico è consistente anche nell’invecchiamento. Volontari anziani (72 ± 1 anno) sono stati randomizzati a ricevere 15 g/d di EAA o 15 g/d di EAA più 3 giorni/settimana di allenamento aerobico supervisionato. Coerentemente con i risultati precedenti, l’assunzione acuta di EAA prima dell’intervento ha aumentato la MPS [78]. Tuttavia, dopo 24 settimane di intervento, il gruppo che combinava EAA ed esercizio aerobico presentava una maggiore risposta sintetica del muscolo agli EAA rispetto al gruppo dei soli EAA [78], indicando che l’esercizio fisico costante sensibilizza ulteriormente il muscolo scheletrico agli effetti anabolici degli EAA. Soprattutto, la maggiore sensibilità del muscolo scheletrico agli EAA si è tradotta in una maggiore qualità muscolare e in una maggiore velocità di cammino sui 400 m nel gruppo EAA più esercizio aerobico [78].
L’esercizio/allenamento ad alta intensità e intermittente (HIIT) e la maggior parte degli sport di squadra (calcio, basket, hockey, tennis, ecc.) richiedono componenti di allenamento sia aerobico che anaerobico/di resistenza. Gli studi condotti sulle proteine suggeriscono che l’esercizio fisico ad alta intensità intrapreso con una maggiore disponibilità di proteine (cioè eseguito in uno stato di alimentazione con proteine) può migliorare sinergicamente l’ipertrofia muscolare [79]. Altri potenziali benefici includono l’aumento della biogenesi mitocondriale, il recupero dell’esercizio, la capacità aerobica e il miglioramento delle prestazioni di sprint [80]. Ad oggi, le poche ricerche che utilizzano EAA e HIIT non sono ancora definitive. In adulti sovrappeso/obesi non allenati, 8 settimane di allenamento a intervalli ad alta intensità (HIIT; 6-10 × 1 min@90% W max: 1 min di riposo) hanno prodotto un aumento significativo delle dimensioni, della sezione trasversale, del volume e della qualità del muscolo della coscia, ma non è stato potenziato sinergicamente da una bassa dose di EAA (3,6 g due volte al giorno) [81]. Inoltre, anche il VO2 è aumentato con l’HIIT, ma non è stato potenziato sinergicamente dagli EAA [82]. Nella stessa coorte, non sono stati riscontrati effetti acuti (3,6 g prima dell’esercizio) o cronici (dopo 4 e 8 settimane di 3,6 g due volte al giorno) dell’integrazione di EAA sul tempo di esaurimento o sulla progressione del carico di lavoro [83]. Sebbene non vi siano prove che suggeriscano che l’integrazione di EAA sia dannosa o attenui gli adattamenti fisiologici associati all’HIIT, non vi sono prove sufficienti per trarre conclusioni sui benefici in termini di adattamento e prestazioni. Tuttavia, c’è motivo di aspettarsi un’interazione degli EAA con l’esercizio aerobico. La sola camminata moderata su tapis roulant (45 minuti al 40% di VO2 di picco) in uomini giovani e anziani ha aumentato la sintesi proteica muscolare subito dopo l’esercizio, con la risposta dei più giovani mantenuta fino a un’ora dopo l’esercizio [84]. Anche la sintesi di fibrinogeno è stata elevata in entrambi i gruppi fino a tre ore dopo l’esercizio [84]. L’apporto di aminoacidi è stato maggiore subito dopo l’esercizio, il che denota ancora una volta l’effetto dell’esercizio sul flusso sanguigno degli arti. Pertanto, se l’aumento del flusso sanguigno si combina con un maggiore apporto di aminoacidi per via orale, ci si aspetta un anabolismo muscolare.
Per quanto riguarda le interazioni con l’esercizio fisico, gli EAA in forma libera possono essere presi in considerazione rispetto alle proteine intatte (siero di latte) sulla base della facilità di assunzione in prossimità e durante l’esercizio. Le formule orali di EAA in forma libera richiedono una digestione minima, comportano un carico gastrico minimo e sono rapidamente assorbite e trasportate in periferia. Per questo motivo, sono ideali per il consumo prima dell’esecuzione di un esercizio fisico rigoroso.
EAA e condizioni cliniche ed esiti
Gli effetti benefici dell’integrazione della dieta con EAA sono stati dimostrati in un’ampia varietà di condizioni cliniche. Le condizioni in gran parte associate all’invecchiamento sono state un obiettivo frequente della terapia con EAA, tra cui la sarcopenia [62,85], le infezioni contratte durante l’assistenza a lungo termine [86], la scarsa funzionalità fisica [63] e l’insufficienza cardiaca [40,87,88]. Gli effetti benefici degli EAA sono stati riportati anche nelle seguenti condizioni o situazioni: riabilitazione [89-92]; ictus [93,94]; riposo a letto/immobilizzazione [8,65,95-97]; malattia arteriosa periferica [98]; insufficienza renale [99-103]; infiammazione [104,105]; malattia critica [106]; cancro del polmone [107]; fibrosi cistica [108]; broncopneumopatia cronica ostruttiva [109-111]; guarigione delle ferite [112]; lesioni cerebrali [113,114]; sindrome metabolica e fattori di rischio cardiovascolare [115-117]; obesità [118,119]; grasso epatico [115,120-122]; e diabete [123-127]. È importante notare che in tutti questi studi gli effetti benefici sono stati osservati nonostante l’assenza di controllo del consumo di EAA contenuti nelle proteine alimentari, il che implica l’importanza di un assorbimento rapido e completo degli EAA liberi in circostanze cliniche in cui la digestione può essere compromessa e la resistenza anabolica è prevalente. I risultati ottenuti nelle popolazioni cliniche evidenziano la necessità e il potenziale impatto dell’integrazione di EAA nell’ambito degli sforzi di riabilitazione da lesioni ortopediche e interventi chirurgici associati. Questi scenari rappresentano gli ambienti clinici più probabili in cui si troveranno gli atleti agonisti e questa rimane un’area di ricerca poco esplorata.
Punti chiave: EAA, esercizio fisico e loro funzione
Gli effetti anabolici degli EAA e dell’esercizio fisico sono interattivi, in quanto la risposta alla combinazione è maggiore della somma delle singole risposte. L’aumento del flusso sanguigno degli arti indotto dall’esercizio contribuisce a questa risposta aumentando il trasporto di EAA nel muscolo scheletrico.
La combinazione di EAA ed esercizio aerobico, così come altre modalità di carico muscolare, è anche anabolizzante per il muscolo scheletrico.
Gli EAA somministrati prima dell’esercizio fisico determinano un anabolismo maggiore rispetto a quelli assunti dopo il completamento dell’esercizio; tuttavia, un effetto anabolico minore può essere realizzato entro un’ora dalla cessazione dell’esercizio.
In assenza di uno stimolo all’esercizio, la somministrazione di EAA in popolazioni anabolicamente resistenti, come quelle affette da invecchiamento e patologie cliniche, si è dimostrata benefica per i risultati clinici ed efficace nel ripristino della forza e delle prestazioni funzionali.
Domande rimanenti
Gli effetti metabolici degli EAA sono stati elegantemente articolati. Inoltre, è stata stabilita la loro interazione metabolica con l’esercizio di resistenza e aerobico. Esiste quindi una base metabolica per la traduzione dell’integrazione di EAA in risultati di performance. Come accennato in precedenza, il sostanziale effetto anabolico dell’integrazione di EAA in forma libera è coerente con la loro efficacia nelle popolazioni che trarrebbero maggior beneficio dal loro utilizzo. Per questo motivo, la stragrande maggioranza dei lavori longitudinali che hanno esaminato l’integrazione di EAA e i risultati funzionali si è svolta in popolazioni anziane o cliniche caratterizzate da insensibilità anabolica muscolare, perdita muscolare e/o debolezza muscolare. Meno lavori hanno documentato gli effetti/risultati a lungo termine associati all’uso di EAA in popolazioni di atleti. Inoltre, sarebbe utile un ulteriore lavoro che definisca il profilo ottimale degli EAA, nonché il dosaggio e la tempistica ottimali per circostanze specifiche.
Stechiometria degli EAA – l’importanza della corretta AA ratio
La stechiometria è il calcolo delle quantità relative di reagenti e prodotti nelle reazioni chimiche. La stechiometria si basa sulla legge di conservazione della massa, secondo la quale la massa totale dei reagenti è uguale alla massa totale dei prodotti, il che porta a capire che le relazioni tra le quantità di reagenti e prodotti formano tipicamente un rapporto di numeri interi positivi. Ciò significa che se le quantità dei singoli reagenti sono note, è possibile calcolare la quantità del prodotto. Al contrario, se un reagente ha una quantità nota e la quantità di prodotto può essere determinata empiricamente, è possibile calcolare anche la quantità degli altri reagenti.
In questo modo si sviluppano metodi per determinare le quantità di composti prodotti o consumati nelle reazioni chimiche e si descrivono alcuni tipi fondamentali di reazioni chimiche. Applicando i concetti e le competenze introdotte, si è in grado di spiegare cosa succede allo zucchero contenuto in una barretta di cioccolato che si mangia, o agli AA contenuti in una fonte proteica e/o integratore, tanto per fare un esempio.
Justus von Liebig
Il concetto di reagente limitante è stato utilizzato dal chimico tedesco Justus von Liebig (1803-1873) nel XIX secolo per ricavare un’importante legge biologica ed ecologica. La legge del minimo di Liebig afferma che la sostanza essenziale disponibile nella quantità più piccola rispetto a un minimo critico controllerà la crescita e la riproduzione di qualsiasi specie vegetale o animale. Quando un gruppo di organismi esaurisce il reagente limitante essenziale, le reazioni chimiche necessarie per la crescita e la riproduzione devono arrestarsi. Vitamine, proteine e altri nutrienti sono essenziali per la crescita del corpo umano e delle popolazioni umane.
Alcuni di voi conosceranno già la “complementarietà degli AA”. In breve, e per fare un esempio, essa riguarda il modo più efficace per ottenere tutti i 9 EAA nella dieta di un vegetariano. La complementarietà degli AA consiste nel combinare due proteine vegetali (ad esempio, legumi e cereali) per ottenere tutti i 9 aminoacidi essenziali per l’organismo. La ripartizione della complementarietà degli AA è la seguente:
Quindi, l’idea della complementarità degli AA, non è altro che l’abbinamento di diversi alimenti della dieta al fine di fornire il giusto equilibrio di aminoacidi per costruire le proteine umane. Le proteine umane richiedono quantità stechiometriche di circa 9 aminoacidi “essenziali”; se ne manca solo uno, la proteina non può essere sintetizzata e ne deriva una malnutrizione proteica. Questo illustra il concetto di “reagente limitante”, ovvero un reagente presente in quantità inferiore a quella stechiometrica rispetto agli altri reagenti.
Un gruppo eterogeneo di culture sopravvissute in tutto il mondo ha adottato diete, per quanto diverse, che forniscono il corretto equilibrio di aminoacidi. Gli antropologi ritengono che l’adozione fortuita di queste diete abbia fornito alle culture un valore di sopravvivenza. Nelle aree in cui il cibo scarseggia, le culture che non adottano diete con un corretto equilibrio di aminoacidi potrebbero non sopravvivere.
In Sud America sono comuni le diete che combinano le tortillas di mais o altri prodotti a base di mais con i fagioli, come la tostada. I fagioli mangiati da soli forniscono quantità limitate di aminoacidi contenenti zolfo, come la metionina e la cisteina, quindi questi aminoacidi limitano la quantità di proteine umane che possono essere sintetizzate. I fagioli contengono grandi quantità di aminoacidi come la lisina e il triptofano, che sono quindi “reagenti in eccesso” quando vengono utilizzati per sintetizzare le proteine umane e vengono degradati in urea e sprecati. Se il grano, il riso o il mais vengono consumati da soli, in genere forniscono quantità di lisina e triptofano che limitano la quantità di proteine umane che possono essere sintetizzate. Ma se i fagioli vengono mangiati insieme ai cereali o al mais, i reagenti in eccesso dei fagioli completano i reagenti limitanti dei cereali (e viceversa). Le proteine umane possono quindi essere sintetizzate in modo efficiente e quantità molto ridotte di aminoacidi vengono semplicemente espulse come urea.
In India, il riso o il chapati vengono consumati con le lenticchie per fornire un equilibrio di aminoacidi.
Per capire meglio il concetto di stechiometria, i questa sede analizzeremo gli esperimenti che riguardano il mais e la malnutrizione proteica che ne può derivare se viene mangiato da solo. È noto che il mais è un alimento povero di proteine di per sé, essendo povero di lisina e triptofano [8], e ancora di più quando viene nixtamalizzato. Queste carenze hanno spinto i ricercatori a sviluppare il QPM (Quality Protein Maze) per aumentare le concentrazioni di questi aminoacidi essenziali nelle sue proteine.
Esperimenti sui ratti, il cui fabbisogno di aminoacidi è simile a quello degli esseri umani, forniscono una base per alcuni calcoli stechiometrici [9]. Come mostra la tabella sottostante, l’aumento di peso è stato minimo quando LYS o TRP sono stati aggiunti separatamente a una dieta “base”. Ciò significa che nessuno dei due, da solo, è un aminoacido limitante e impedisce la sintesi delle proteine dei topi. Ma quando sono stati aggiunti entrambi, è stato misurato un aumento significativo, insieme a una diminuzione dell’urea sierica. Ciò significa che entrambi erano limitanti e che, quando sono stati aggiunti entrambi, è stata sprecata una quantità molto minore di aminoacidi. Gli aminoacidi sono stati destinati alla sintesi proteica, anziché essere semplicemente metabolizzati ed escreti come urea. Mentre l’aggiunta di isoleucina nella dieta 5 ha fatto poca differenza (quindi deve essere fornita adeguatamente dal mais), l’aggiunta della sola treonina (THR) alla dieta 4 ha aumentato l’aumento di peso corporeo, quindi deve essere limitante in una dieta di mais + LYS + TRP. L’aggiunta di ILE, metionina (MET), istidina (HIS) e valina (VAL) costituisce infine una dieta quasi bilanciata, come dimostrano i grandi aumenti di peso e la bassa urea sierica della dieta 8.
La dieta di base consisteva in 920,2 g di mais, 30,0 g di olio di mais, 35,0 g di miscela di minerali, 10 g di miscela di vitamine, 2,5 g di calcare e 2,3 g di colina. b Questo forte aumento è dovuto al raddoppio delle proteine totali della dieta, piuttosto che all’inefficienza proteica.
Lo stesso documento[10] fornisce la sequenza, dalla più limitante alla meno, degli aminoacidi del mais e il fabbisogno del ratto (simile a quello umano) pubblicato dal NRC[11]. Abbiamo aggiunto le quantità di aminoacidi nelle lenticchie per rappresentare i fagioli[12].
Corretta stechiometria o ratio degli aminoacidi:
In base ai dati della Tabella II, calcolare il rapporto molare ottimale tra lisina (LYS, C6H14N2O2, massa molare 146,19 g/mol) e arginina (ARG, C6H14N4O2, massa molare 174,2 g/mol) nella dieta.
Aminoacido limitante:
Dai dati della tabella precedente, calcolare quale, LYS o ARG, è il reagente limitante in una dieta a base di mais?
c. Quanto aminoacido in eccesso verrà sprecato in 1kg di dieta a base di mais?
Soluzione
a. L’equazione bilanciata avrà dei coefficienti che dobbiamo determinare:
x LYS + y ARG + altri amminoacidi → Proteina + acqua
Secondo la teoria atomica, per ogni y mole di ARG sono necessarie x mol di LYS per produrre proteine utili. Se la dieta è ottimale quando si consumano 7,0 g di LYS per ogni 6,0 g di ARG (questo rapporto potrebbe essere fornito dalle uova, per esempio), possiamo calcolare il rapporto stechiometrico ottimale:
Questo rapporto esatto di aminoacidi potrebbe non essere presente in una particolare proteina del ratto, ma è il rapporto medio per tutte le proteine del ratto. L’equazione chimica (parziale) è
b. Vediamo qual è il rapporto molare nella dieta a base di mais.
Chiaramente, la LYS è l’amminoacido limitante perché il rapporto iniziale tra LYS e ARG (0,55:1) è molto inferiore al rapporto stechiometrico richiesto (1,4:1).
Dall’esempio della dieta a base di mais si può iniziare a capire cosa bisogna fare per determinare quale dei due reagenti, X o Y, è limitante. Dobbiamo confrontare il rapporto stechiometrico S(X/Y) con il rapporto effettivo delle quantità di X e Y inizialmente mescolate.
La regola generale corrispondente, per qualsiasi reagente X e Y, è
Naturalmente, quando le quantità di X e Y sono esattamente nel rapporto stechiometrico, entrambi i reagenti vengono consumati completamente nello stesso momento e nessuno dei due è in eccesso.
Possiamo verificare che LYS è limitante calcolando la quantità di LYS che sarebbe necessaria se tutta la ARG reagisse, utilizzando un rapporto stechiometrico:
Allo stesso modo, se tutta la LYS reagisce, la quantità di ARG richiesta è
Questa quantità è molto inferiore a quella presente, quindi ARG è il reagente in eccesso.
c. Possiamo facilmente utilizzare le masse molari per convertire le quantità di ciascun amminoacido in masse.
Prepariamo una tabella:
Al termine della reazione, rimangono 3,14 g dei 5,0 g originali (63%), che saranno metabolizzati in urea ed escreti. Dell’intera dieta di 2,4 g di LYS + 5,0 g di ARG, vediamo che 3,14 g o il 42% viene sprecato. Che spreco di cibo e di risorse per produrlo! Si noti che anche la porzione di mais dovrà essere maggiore rispetto alla porzione di dieta ottimale per ottenere la stessa quantità di proteine, perché il mais ha solo 5 g/kg di ARG, mentre la dieta ottimale ha 6,0 g/kg. La dieta ottimale potrebbe essere fornita dalla carne o dalle uova, ma sono impegnative per l’ambiente e presentano problemi di salute.
Una dieta di sussistenza a base di fagioli può anche portare a una malnutrizione proteica, come mostra la tabella II. Sebbene il contenuto di lisina sia molto superiore a quello dei fagioli, i livelli di metionina (MET) e cistina (CYS) sono bassi, così come quelli di fenilalanina (PHE) e tirosina (TYR).
Ma supponiamo che fagioli e mais vengano consumati nello stesso giorno, in una dieta che mescola in egual misura mais e lenticchie. Ricalcolate ora l’amminoacido limitante in una dieta con 2,4 + 6,3 = 8,7 g di LYS e 5,0 + 6,97 = 11,97 g di ARG.
b. In questo caso, quanta parte dell’amminoacido in eccesso viene sprecata?
Soluzione
Ora vediamo che il rapporto tra le quantità presenti è
0,060 mol LYS / 0,069 mol ARG = 0,869,
che è ancora inferiore al rapporto stechiometrico,
7 mol LYS / 5 mol ARG = 1,4
Quindi LYS è ancora una volta l’amminoacido limitante. Ricalcolando i valori in una tabella come sopra, otteniamo
In questo caso, da una porzione di 11,97 g rimangono 4,61 g di ARG in eccesso, ovvero il 38% dell’ARG. La dieta totale è di 8,7 g di LYS + 11,97 g di ARG = 20,7 g, di cui solo il 22% viene sprecato, un grande miglioramento rispetto al caso del solo mais di cui sopra.
Per progettare una dieta adeguata, gli alimenti proteici (o singoli AA) complementari dovrebbero essere scelti dalle tabelle dei contenuti di aminoacidi degli alimenti.
Il processo di Nixtamalizzazione converte parte del già limitante triptofano in 2-amminoacetofenone, aggravando lo scarso valore nutrizionale e di sopravvivenza del mais. Ma la Nixtamalizzazione rende anche più biodisponibile la Niacina contenuta nei chicchi, riducendo l’incidenza della Pellagra e più che compensando la perdita di aminoacidi se nella dieta sono inclusi anche molti fagioli. La dieta a base di fagioli e mais, anche con la Nixtamalizzazione, ha un valore di sopravvivenza.
Ninidrina
Per rilevare un amminoacido (anche in un’impronta digitale nella chimica forense), si utilizza spesso il test della ninidrina.
Nel test della ninidrina, due molecole di ninidrina (C9H6O4, mostrato a sinistra) vengono legate dalla N attaccata al primo carbonio della catena aminoacidica, producendo lo ione blu/viola mostrato di seguito.
Se si utilizzano 2,00 mg di ninidrina (Nin) per rilevare 2 mg di TRP, è stata aggiunta abbastanza ninidrina da reagire con tutta il TRP? Qual è il reagente limitante e quale massa di H2O si formerà?
La soluzione
Il rapporto stechiometrico che collega Nin e TRP è
b) La quantità di prodotto acquoso che si forma in una reazione può essere calcolata attraverso un appropriato rapporto stechiometrico dalla quantità di un reagente consumato. Rimarrà una parte del reagente in eccesso TRP, ma tutta la quantità iniziale di Nin sarà consumata. Pertanto, utilizziamo nNin (iniziale) per calcolare la quantità di H2O ottenuta.
Si tratta di 0,302mg di acqua.
Questi calcoli possono essere organizzati come una tabella, con le voci sotto i rispettivi reagenti e prodotti dell’equazione chimica. I calcoli sono mostrati per ogni possibile caso, ipotizzando che un reagente sia completamente consumato e determinando se è presente una quantità sufficiente di altri reagenti per consumarlo. In caso contrario, lo scenario viene scartato.
Come si può notare dall’esempio, nel caso in cui vi sia un reagente limitante, per calcolare la quantità di prodotto formato si deve utilizzare la quantità iniziale del reagente limitante. Utilizzare la quantità iniziale di un reagente presente in eccesso non sarebbe corretto, perché tale reagente non viene consumato interamente.
Sulla traccia della “complementarietà AA” potremmo prendere come esempio i piselli che sono poveri di Metionina ma ricchi di Lisina e, al contrario, il riso integrale che è ricco di Metionina ma povero di Lisina.
Se dovessimo prendere come esempio principale al quale applicare una stechiometria con l’aggiunta del reagente limitante da AA integrativi.
Vediamo dalla tabella sopra esposta che il reagente limitante nelle proteine del pisello è la Metionina mentre il reagente in eccesso è la Lisina.
In base ai dati precedentemente riportati, calcolare il rapporto molare ottimale tra Metionina (MHT, C5H11NO2S, massa molare 149,21 g/mol) e Lisina (LYS, C6H14N2O2, massa molare 146,19 g/mol) nella dieta.
Quindi, procedendo rapidamente…
x MHT + y LYS + altri amminoacidi → Proteina + acqua
Secondo la teoria atomica, per ogni y mole di LYS sono necessarie x mol di MHT per produrre proteine utili. Se la dieta è ottimale quando si consumano 7,0 g di LYS per ogni 2,1 g di MHT (questo rapporto potrebbe essere fornito dalle uova, per esempio), possiamo calcolare il rapporto stechiometrico ottimale:
Accelerando la procedura di calcolo, sempre sulla linea prima esposta, abbiamo 1.5g di MHT e 11.42g di LYS con mole rispettivamente di 149.21 e 146.19 = 0.010 MHT e 0.078 LYS = 0.13/1
Anche qui, la MHT è l’amminoacido limitante perché il rapporto iniziale tra MHT e LYS (0,13:1) è inferiore al rapporto stechiometrico richiesto (0.29:1).
Di conseguenza, per ogni 100g di proteine del pisello andrebbero addizionati 140mg di MHT al fine di migliorare la ratio stechiometrica.
Nel riso “bianco”, comunemente consumato dai Bodybuilder, abbiamo la presenza dello stesso reagente limitante del pisello; ossia la MHT con il reagente in eccesso LYS.
Punto della situazione
Dovremmo aver compreso, ora, che un prodotto contenete EAA andrebbe valutato in base alla sua formulazione stechiometrica la quale, per essere considerata ottimale, non deve presentare ne carenze ne eccessi per uno o più dei 9 EAA.
Sul mercato attuale sono presenti poche formulazioni stechiometriche di EEA valide. Tra queste vi è certamente la MAP® . Il MAP [Master Amino Acid Pattern®], è una combinazione stechiometrica di EAA usata anche in campo clinico. MAP fornisce, con il minor peso e volume, il maggior valore nutritivo proteico/AA in assoluto.
Clinicamente, il MAP è particolarmente consigliabile per coloro che soffrono di nausea o di inappetenza sia per ragioni fisio-patologiche (AIDS, cancro, anemia, denutrizione, ecc.) sia per ragioni psicologiche (anoressia, bulimia, ecc.)
Il MAP fornisce, in soli 10g, un valore nutritivo proteico/AA pari a circa 350g di carne rossa, pesce o pollame, con un valore energetico di sole 50Kcal. Per via della sua stechiometria, 10g di MAP, a differenza dei 350g di carne, pesce o pollame a cui equivale, rilascia una quantità minima (meno dell’1%) di cataboliti azotati. Quindi, il MAP, o altra formulazione stechiometricamente corretta, è particolarmente consigliabile per l’alimentazione di coloro che hanno deficit di funzionalità epatica o renale o per coloro i quali desiderano gravarne il meno possibile per via del già presente impatto stressorio d’organo dato da terapia iatrogena.
Il MAP viene completamente assorbito in meno di 23 minuti dalla sua ingestione, poiché è assimilabile ad una proteina alimentare digerita (ossia scomposta nelle sue singole unità AA). Il MAP non necessita quindi dell’azione dell’ enzima peptidasi, ed il suo assorbimento, stimola la secrezione biliare, pancreatica ed intestinale in misura minima. Quindi, il MAP viene particolarmente consigliato per tutti coloro che soffrono di disturbi gastrointestinali anche per bisogno di consumare grosse quantità di cibo [vedi bodybuilder].
Punti chiave del MAP:
Il MAP fornisce un 99% di Net Nitrogen Utilization (NNU): vale a dire che il 99% dei suoi aminoacidi costitutivi agiscono come precursori della sintesi proteica corporea.
Il MAP fornisce una quantità minima (meno dell’ 1%) di cataboliti azotati.
Il MAP é digerito e assorbito in meno di 23 minuti.
Sintesi e conclusioni finali:
I 15 punti seguenti costituiscono al momento “le linee guida” sugli EEA avallati dalla ISSN:
L’integrazione di EAA in forma libera (non derivati da proteine esogene intatte) è un robusto stimolatore della sintesi e del turnover delle proteine muscolari.
Gli EAA stimolano la sintesi proteica muscolare più di un isolato proteico isonitrogeno.
L’ingestione di EAA produce un rapido aumento delle concentrazioni periferiche e il trasporto degli aminoacidi nel muscolo scheletrico.
La stimolazione degli EAA della sintesi proteica muscolare può avvenire con dosaggi multipli e non interferisce con gli effetti dei pasti.
Singoli o gruppi di EAA possono avviare il processo di stimolazione; tuttavia, una stimolazione significativa e prolungata si verifica quando vengono consumati tutti gli EAA.
La stimolazione della sintesi proteica a riposo da parte degli EAA avviene con dosaggi che vanno da 1,5 g a 18 g.
Una percentuale maggiore di leucina (%/g) contenuta nelle composizioni di EAA ingerite è necessaria per stimolare al massimo la sintesi proteica muscolare nelle popolazioni (invecchiamento, patologie cliniche) che dimostrano resistenza anabolica.
Nelle popolazioni anabolicamente resistenti, l’integrazione longitudinale di EAA migliora i risultati funzionali.
Gli effetti degli EAA e dell’esercizio fisico sono interattivi, tanto da amplificare gli effetti combinati. Questa interazione è dovuta a un maggiore apporto di EAA al muscolo in esercizio, grazie all’aumento del flusso sanguigno e alle più alte concentrazioni di EAA nel sangue.
Le risposte anaboliche sono costantemente riportate con la combinazione dell’ingestione di EAA con l’esercizio di resistenza o aerobico. Questo effetto si mantiene con l’invecchiamento.
L’integrazione di EAA in forma libera rientra nel limite massimo di sicurezza del consumo giornaliero abituale.
L’integrazione di EAA è efficace nella maggior parte degli studi clinici e delle condizioni.
Numerosi studi longitudinali che prevedono l’integrazione di EAA in popolazioni anziane riportano costantemente miglioramenti favorevoli sia a livello metabolico che funzionale.
Sono necessarie ulteriori ricerche per esaminare il potenziale impatto della somministrazione di EAA in popolazioni atletiche sottoposte intenzionalmente o meno a privazione energetica sui cambiamenti del metabolismo proteico muscolare e sui cambiamenti associati in termini di prestazioni e composizione corporea.
Sono necessarie ulteriori ricerche per esaminare il ruolo della somministrazione di EAA a popolazioni di atleti che attraversano periodi inaspettati e improvvisi di inattività, probabilmente secondari a lesioni acute e a periodi di riabilitazione che seguono abitualmente interventi chirurgici.
L’integrazione con EAA vede le sue migliori applicazioni in:
Presenza di una carenza proteica, o comunque una condizione alimentare che non da modo di apportare la giusta quantità di EAA con gli alimenti;
Bodybuilder in ipercalorica che vogliono ridurre il carico di cibo e massimizzare al meglio digestione e assorbimento AA.
Soggetto che non tollera le proteine in polvere;
Inserimento “compensativo” per ridurre il consumo di fonti proteiche intere.
Soggetto con problematiche digestive di base.
Assicuratevi di acquistare prodotti di alta qualità altrimenti i vantaggi ottenibili e sopra esposti non si verificheranno.
Parliamo comunque di un integratore “compensativo” e “migliorativo” dello stato e compliance alimentare del utilizzatore.
Gli EEA non sostituiscono tutti i 21 AA; utilizzarli come substrato di sintesi per la matrice mancante è uno spreco di risorse e di soldi. Dovreste aver capito che è tutta una questione di equilibrio funzionale.
1. Bradford, H, Schmidt, V.. The history of the discovery of the amino acids. Chem Rev. 1931;9:169–864. doi: 10.1021/cr60033a001 [CrossRef] [Google Scholar]
2. Dietary reference intakes for energy, carbohydrates, fiber, fat, protein and amino acids (macronutrients). Washington, DC: Institute of Medicine of the National Academies, 2005. 2002/2005. Report No. [Google Scholar]
3. Elango, R, Ball, RO, Pencharz, PB. Recent advances in determining protein and amino acid requirements in humans. British Journal Of Nutrition. 2012;108(2):S22–30. Epub 2012/10/31. PubMed PMID: 23107531. doi: 10.1017/S0007114512002504 [PubMed] [CrossRef] [Google Scholar]
4. Food and Agriculture Organization of the United Nations Dietary protein quality evaluation in human nutrition. Rome: FAO Food and Nutrition. 2013. 31 March – 2 April 2011. Report No. [Google Scholar]
5. CE, B, JS, A, DT, H. Protein quality in humans: assessment and in vitro estimation. Westport Connecticut: AVI Publishing, Inc; 1981. [Google Scholar]
6. Bifari, F, Ruocco, C, Decimo, I, et al. Amino acid supplements and metabolic health: a potential interplay between intestinal microbiota and systems control. Genes Nutr. 2017;12:27. Epub 2017/10/19. PubMed PMID: 29043007; PMCID: PMC5628494. doi: 10.1186/s12263-017-0582-2 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
7. Paddon-Jones, D, Sheffield-Moore, M, Urban, RJ, et al. Essential amino acid and carbohydrate supplementation ameliorates muscle protein loss in humans during 28 days bedrest. J Clin Endocrinol Metab. 2004;89(9):4351–4358. PubMed PMID: 15356032. doi: 10.1210/jc.2003-032159 [PubMed] [CrossRef] [Google Scholar]
8. Fitts, RH, Romatowski, JG, Peters, JR, et al. The deleterious effects of bed rest on human skeletal muscle fibers are exacerbated by hypercortisolemia and ameliorated by dietary supplementation. Am J Physiol Cell Physiol. 2007;293(1):C313–20. PubMed PMID: 17409123. doi: 10.1152/ajpcell.00573.2006 [PubMed] [CrossRef] [Google Scholar]
9. Biolo, G, Tipton, KD, Klein, S, et al. An abundant supply of amino acids enhances the metabolic effect of exercise on muscle protein. Am J Physiol. 1997;273(1 Pt 1):E122–9. PubMed PMID: 9252488. doi: 10.1152/ajpendo.1997.273.1.E122. [PubMed] [CrossRef] [Google Scholar]
10. Magaway, C, Kim, E, Jacinto, E. Targeting mTOR and metabolism in cancer: lessons and innovations. Cells. 2019;8(12). Epub 2019/12/11. PubMed PMID: 31817676; PMCID: PMC6952948. doi: 10.3390/cells8121584 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
15. Drummond, MJ, Rasmussen, BB. Leucine-enriched nutrients and the regulation of mammalian target of rapamycin signalling and human skeletal muscle protein synthesis. Current opinion in clinical nutrition and metabolic care. 2008;11(3):222–226. Epub 2008/04/12. PubMed PMID: 18403916; PMCID: PMC5096790. doi: 10.1097/MCO.0b013e3282fa17fb [PMC free article] [PubMed] [CrossRef] [Google Scholar]
16. Dreyer, HC, Fujita, S, Glynn, EL, et al. Resistance exercise increases leg muscle protein synthesis and mTOR signalling independent of sex. Acta Physiol (Oxf). 2010;199(1):71–81. Epub 2010/01/15. PubMed PMID: 20070283; PMCID: PMC2881180. doi: 10.1111/j.1748-1716.2010.02074.x [PMC free article] [PubMed] [CrossRef] [Google Scholar]
17. Maier, T, Guell, M, Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583(24):3966–3973. Epub 2009/10/24. PubMed PMID: 19850042. doi: 10.1016/j.febslet.2009.10.036. [PubMed] [CrossRef] [Google Scholar]
18. Greenhaff, PL, Karagounis, LG, Peirce, N, et al. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am J Physiol. 2008;295(3):E595–604. PubMed PMID: 18577697. doi: 10.1152/ajpendo.90411.2008 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
19. Crick, FH. On protein synthesis. Symp Soc Exp Biol. 1958;12:138–163. Epub 1958/01/01. PubMed PMID: 13580867. [PubMed] [Google Scholar]
20. Graber, TG, Borack, MS, Reidy, PT, et al. Essential amino acid ingestion alters expression of genes associated with amino acid sensing, transport, and mTORC1 regulation in human skeletal muscle. Nutri Metabol. 2017;14:35. Epub 2017/05/16. PubMed PMID: 28503190; PMCID: PMC5426042. doi: 10.1186/s12986-017-0187-1 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
21. Wolfe, RR, Chinkes, D. Isotope tracers in metabolic research: principles and practice of kinetic analysis. New York, New Yor: Wiley-Liss; 2004. p. 274. [Google Scholar]
22. Biolo, G, Chinkes, D, Zhang, X, et al. Harry M. Vars research award: a new model to determine in vivo the relationship between amino acid transmembrane transport and protein kinetics in muscle. JPEN J Parenter Enteral Nutr. 1992;16(4):305–315. doi: 10.1177/0148607192016004305 [PubMed] [CrossRef] [Google Scholar]
23. Biolo, G, Fleming, RY, Maggi, SP, et al. Transmembrane transport and intracellular kinetics of amino acids in human skeletal muscle. Am J Physiol. 1995;268(1 Pt 1):E75–84. PubMed PMID: 7840186. doi: 10.1152/ajpendo.1995.268.1.E75. [PubMed] [CrossRef] [Google Scholar]
24. Tipton, KD, Borsheim, E, Wolf, SE, et al. Acute response of net muscle protein balance reflects 24-h balance after exercise and amino acid ingestion. Am J Physiol (Endocrinol Metab). 2003;284(1):E76–89. PubMed PMID: 12388164. doi: 10.1152/ajpendo.00234.2002. [PubMed] [CrossRef] [Google Scholar]
25. Plotkin, DL, Delcastillo, K, Van Every, DW, et al. Isolated leucine and branched-chain amino acid supplementation for enhancing muscular strength and hypertrophy: a narrative review. Int J Sport Nutr Exercise Metab. 2021;31(3):292–301. Epub 2021/03/21. PubMed PMID: 33741748. doi: 10.1123/ijsnem.2020-0356 [PubMed] [CrossRef] [Google Scholar]
26. Fuchs, CJ, Hermans, WJH, Holwerda, AM, et al. Branched-chain amino acid and branched-chain ketoacid ingestion increases muscle protein synthesis rates in vivo in older adults: a double-blind, randomized trial. Am J Clin Nutr. 2019;110(4):862–872. Epub 2019/06/30. PubMed PMID: 31250889; PMCID: PMC6766442. doi: 10.1093/ajcn/nqz120. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
27. Pinals, RS, Harris, ED, Burnett, JB, et al. Treatment of rheumatoid arthritis with L-histidine: a randomized, placebo-controlled, double-blind trial. J Rheumatol. 1977;4(4):414–419. Epub 1977/01/01. PubMed PMID: 342692. [PubMed] [Google Scholar]
28. Jackman, SR, Witard, OC, Jeukendrup, AE, et al. Branched-chain amino acid ingestion can ameliorate soreness from eccentric exercise. Med & Sci In Sports & Ex. 2010;42(5):962–970. PubMed PMID: 19997002. doi: 10.1249/MSS.0b013e3181c1b798 [PubMed] [CrossRef] [Google Scholar]
29. Cynober, L, Bier, DM, Kadowaki, M, et al. A proposal for an upper limit of leucine safe intake in healthy adults. J Nutr. 2012;142(12):2249S–2250S. Epub 2012/10/26. PubMed PMID: 23096009. doi: 10.3945/jn.112.160853 [PubMed] [CrossRef] [Google Scholar]
30. Kim, IY, Williams, RH, Schutzler, SE, et al. Acute lysine supplementation does not improve hepatic or peripheral insulin sensitivity in older, overweight individuals. Nutri Metabol. 2014;11(1):49. Epub 2014/10/18. PubMed PMID: 25324894; PMCID: PMC4198625. doi: 10.1186/1743-7075-11-49 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
31. Report of the expert advisory committee on amino acids. Ottawa, Canada: Minister of Supply and Services Canada, 1990. [Google Scholar]
32. Ryan-Harshman, M, Leiter, LA, Anderson, GH. Phenylalanine and aspartame fail to alter feeding behavior, mood and arousal in men. Physiol Behav. 1987;39(2):247–253. Epub 1987/01/01. PubMed PMID: 3575461. doi: 10.1016/0031-9384(87)90017-5. [PubMed] [CrossRef] [Google Scholar]
33. Growdon, JH, Nader, TM, Schoenfeld, J, et al. L-threonine in the treatment of spasticity. Clin Neuropharmacol. 1991;14(5):403–412. Epub 1991/10/01. PubMed PMID: 1742749. doi: 10.1097/00002826-199110000-00003 [PubMed] [CrossRef] [Google Scholar]
34. Cynober, L, Bier, DM, Kadowaki, M, et al. Proposals for upper limits of safe intake for arginine and tryptophan in young adults and an upper limit of safe intake for leucine in the elderly. J Nutr. 2016;146(12):2652S–2654S. Epub 2016/12/10. PubMed PMID: 27934658. doi: 10.3945/jn.115.228478 [PubMed] [CrossRef] [Google Scholar]
35. Volpi, E, Kobayashi, H, Sheffield-Moore, M, et al. Essential amino acids are primarily responsible for the amino acid stimulation of muscle protein anabolism in healthy elderly adults. Am J Clin Nutr. 2003;78(2):250–258. PubMed PMID: 12885705. doi: 10.1093/ajcn/78.2.250 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
36. Kobayashi, H, Borsheim, E, Anthony, TG, et al. Reduced amino acid availability inhibits muscle protein synthesis and decreases activity of initiation factor eIF2B. Am J Physiol. 2003;284(3):E488–98. PubMed PMID: 12556349. doi: 10.1152/ajpendo.00094.2002 [PubMed] [CrossRef] [Google Scholar]
37. Wilkinson, DJ, Bukhari, SSI, Phillips, BE, et al. Effects of leucine-enriched essential amino acid and whey protein bolus dosing upon skeletal muscle protein synthesis at rest and after exercise in older women. Clin Nutr. 2018;37(6 Pt A):2011–2021. Epub 2017/10/17. PubMed PMID: 29031484; PMCID: PMC6295981. doi: 10.1016/j.clnu.2017.09.008 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
38. Tipton, KD, Gurkin, BE, Matin, S, et al. Nonessential amino acids are not necessary to stimulate net muscle protein synthesis in healthy volunteers. J Nutr Biochem. 1999;10(2):89–95. PubMed PMID: 15539275. doi: 10.1016/S0955-2863(98)00087-4. [PubMed] [CrossRef] [Google Scholar]
39. Paddon-Jones, D, Sheffield-Moore, M, Katsanos, CS, et al. Differential stimulation of muscle protein synthesis in elderly humans following isocaloric ingestion of amino acids or whey protein. Exp Gerontol. 2006;41(2):215–219. PubMed PMID: 16310330. doi: 10.1016/j.exger.2005.10.006 [PubMed] [CrossRef] [Google Scholar]
40. Kim, IY, Park, S, Smeets, E, et al. Consumption of a specially-formulated mixture of essential amino acids promotes gain in whole-body protein to a greater extent than a complete meal replacement in older women with heart failure. Nutrients. 2019;11(6). Epub 2019/06/20. PubMed PMID: 31212940; PMCID: PMC6627910. doi: 10.3390/nu11061360 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
41. Bukhari, SS, Phillips, BE, Wilkinson, DJ, et al. Intake of low-dose leucine-rich essential amino acids stimulates muscle anabolism equivalently to bolus whey protein in older women at rest and after exercise. Am J Physiol. 2015;308(12):E1056–65. Epub 2015/04/02. PubMed PMID: 25827594. doi: 10.1152/ajpendo.00481.2014 [PubMed] [CrossRef] [Google Scholar]
42. Park, S, Church, DD, Azhar, G, et al. Anabolic response to essential amino acid plus whey protein composition is greater than whey protein alone in young healthy adults. J Int Soc Sports Nutr. 2020;17(1):9. Epub 2020/02/12. PubMed PMID: 32041644; PMCID: PMC7011510. doi: 10.1186/s12970-020-0340-5 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
43. Church, DD, Hirsch, KR, Park, S, et al. Essential amino acids and protein synthesis: insights into maximizing the muscle and whole-body response to feeding. Nutrients. 2020;12(12). Epub 2020/12/06. PubMed PMID: 33276485; PMCID: PMC7760188. doi: 10.3390/nu12123717 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
44. Adibi, SA, Fogel, MR, Agrawal, RM. Comparison of free amino acid and dipeptide absorption in the jejunum of sprue patients. Gastroenterology. 1974;67(4):586–591. Epub 1974/10/01. PubMed PMID: 4412534. doi: 10.1016/S0016-5085(19)32783-0. [PubMed] [CrossRef] [Google Scholar]
45. Drummond, MJ, Glynn, EL, Fry, CS, et al. An increase in essential amino acid availability upregulates amino acid transporter expression in human skeletal muscle. Am J Physiol. 2010;298(5):E1011–8. Epub 2010/03/23. PubMed PMID: 20304764; PMCID: PMC2867366. doi: 10.1152/ajpendo.00690.2009 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
46. Bohe, J, Low, A, Wolfe, RR, et al. Human muscle protein synthesis is modulated by extracellular, not intramuscular amino acid availability: a dose-response study. Journal Of Physiology. 2003;552(Pt 1):315–324. PubMed PMID: 12909668. doi: 10.1113/jphysiol.2003.050674. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
47. Mitchell, WK, Phillips, BE, Williams, JP, et al. A dose- rather than delivery profile-dependent mechanism regulates the “muscle-full” effect in response to oral essential amino acid intake in young men. J Nutr. 2015;145(2):207–214. Epub 2015/02/04. PubMed PMID: 25644339; PMCID: PMC4304023. doi: 10.3945/jn.114.199604 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
48. Anderson, GH, Moore, SE. Dietary proteins in the regulation of food intake and body weight in humans. J Nutr. 2004;134(4):974S–979S. PubMed PMID: 15051857. doi: 10.1093/jn/134.4.974S. [PubMed] [CrossRef] [Google Scholar]
49. Paddon-Jones, D, Sheffield-Moore, M, Aarsland, A, et al. Exogenous amino acids stimulate human muscle anabolism without interfering with the response to mixed meal ingestion. Am J Physiol. 2005;288(4):E761–7. PubMed PMID: 15572657. doi: 10.1152/ajpendo.00291.2004 [PubMed] [CrossRef] [Google Scholar]
50. Abdulla, H, Bass, JJ, Stokes, T, et al. The effect of oral essential amino acids on incretin hormone production in youth and ageing. Endocrinol Diabetes Metab. 2019;2(4):e00085. Epub 2019/10/09. PubMed PMID: 31592446; PMCID: PMC6775449. doi: 10.1002/edm2.85 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
51. Park, S, Jang, J, Choi, MD, et al. The anabolic response to dietary protein is not limited by the maximal stimulation of protein synthesis in healthy older adults: a randomized crossover trial. Nutrients. 2020;12(11). Epub 2020/10/30. PubMed PMID: 33114585; PMCID: PMC7693481. doi: 10.3390/nu12113276 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
52. Fry, CS, Drummond, MJ, Glynn, EL, et al. Aging impairs contraction-induced human skeletal muscle mTORC1 signaling and protein synthesis. Skelet Muscle. 2011;1(1):11. Epub 2011/07/30. PubMed PMID: 21798089; PMCID: PMC3156634. doi: 10.1186/2044-5040-1-11 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
53. Katsanos, CS, Kobayashi, H, Sheffield-Moore, M, et al. Aging is associated with diminished accretion of muscle proteins after the ingestion of a small bolus of essential amino acids. Am J Clin Nutr. 2005;82(5):1065–1073. PubMed PMID: 16280440. doi: 10.1093/ajcn/82.5.1065 [PubMed] [CrossRef] [Google Scholar]
54. Paddon-Jones, D, Sheffield-Moore, M, Zhang, XJ, et al. Amino acid ingestion improves muscle protein synthesis in the young and elderly. Am J Physiol. 2004;286(3):E321–8. PubMed PMID: 14583440. doi: 10.1152/ajpendo.00368.2003 [PubMed] [CrossRef] [Google Scholar]
55. Katsanos, CS, Kobayashi, H, Sheffield-Moore, M, et al. A high proportion of leucine is required for optimal stimulation of the rate of muscle protein synthesis by essential amino acids in the elderly. Am J Physiol. 2006;291(2):E381–7. PubMed PMID: 16507602. doi: 10.1152/ajpendo.00488.2005 [PubMed] [CrossRef] [Google Scholar]
56. Carbone, JW, McClung, JP, Pasiakos, SM. Skeletal muscle responses to negative energy balance: effects of dietary protein. Adv nutri (Bethesda, Md). 2012;3(2):119–126. Epub 2012/04/21. PubMed PMID: 22516719; PMCID: PMC3648712 conflicts of interest. doi: 10.3945/an.111.001792 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
57. Trappe, TA, Gastaldelli, A, Jozsi, AC, et al. Energy expenditure of swimmers during high volume training. Medi and sci sports and exerci. 1997;29(7):950–954. Epub 1997/07/01. PubMed PMID: 9243495. doi: 10.1097/00005768-199707000-00015 [PubMed] [CrossRef] [Google Scholar]
58. Gwin, JA, Church, DD, Hatch-McChesney, A, et al. Effects of high versus standard essential amino acid intakes on whole-body protein turnover and mixed muscle protein synthesis during energy deficit: a randomized, crossover study. Clin Nutr. 2021;40(3):767–777. Epub 2020/08/10. PubMed PMID: 32768315. doi: 10.1016/j.clnu.2020.07.019 [PubMed] [CrossRef] [Google Scholar]
59. Biolo, G, Fleming, RY, Maggi, SP, et al. Inverse regulation of protein turnover and amino acid transport in skeletal muscle of hypercatabolic patients. J Clin Endocrinol Metab. 2002;87(7):3378–3384. PubMed PMID: 12107253. doi: 10.1210/jcem.87.7.8699 [PubMed] [CrossRef] [Google Scholar]
60. Dillon, EL, Sheffield-Moore, M, Paddon-Jones, D, et al. Amino acid supplementation increases lean body mass, basal muscle protein synthesis, and insulin-like growth factor-I expression in older women. J Clin Endocrinol Metab. 2009;94(5):1630–1637. PubMed PMID: 19208731. doi: 10.1210/jc.2008-1564 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
61. Borsheim, E, Bui, QU, Tissier, S, et al. Effect of amino acid supplementation on muscle mass, strength and physical function in elderly. Clin Nutr. 2008;27(2):189–195. PubMed PMID: 18294740. doi: 10.1016/j.clnu.2008.01.001. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
62. Kim, HK, Suzuki, T, Saito, K, et al. Effects of exercise and amino acid supplementation on body composition and physical function in community-dwelling elderly Japanese sarcopenic women: a randomized controlled trial. J Am Geriatr Soc. 2012;60(1):16–23. PubMed PMID: 22142410. doi: 10.1111/j.1532-5415.2011.03776.x [PubMed] [CrossRef] [Google Scholar]
63. Azhar, G, Wei, JY, Schutzler, SE, et al. Daily consumption of a specially formulated essential amino acid-based dietary supplement improves physical performance in older adults with low physical functioning. J Gerontol A Biol Sci Med Sci. 2021;76(7):1184–1191. Epub 2021/01/22. PubMed PMID: 33475727; PMCID: PMC8202157. doi: 10.1093/gerona/glab019 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
64. Hwang, CL, Chien, CL, Wu, YT. Resistance training increases 6-minute walk distance in people with chronic heart failure: a systematic review. J Physiother. 2010;56(2):87–96. Epub 2010/05/21. PubMed PMID: 20482475. doi: 10.1016/s1836-9553(10)70038-2. [PubMed] [CrossRef] [Google Scholar]
65. Ferrando, AA, Paddon-Jones, D, Hays, NP, et al. EAA supplementation to increase nitrogen intake improves muscle function during bed rest in the elderly. Clin Nutr. 2010;29:18–23. PubMed PMID: 19419806. doi: 10.1016/j.clnu.2009.03.009. [PubMed] [CrossRef] [Google Scholar]
66. Biolo, G, Maggi, SP, Williams, BD, et al. Increased rates of muscle protein turnover and amino acid transport after resistance exercise in humans. Am J Physiol. 1995;268(3 Pt 1):E514–20. PubMed PMID: 7900797. doi: 10.1152/ajpendo.1995.268.3.E514. [PubMed] [CrossRef] [Google Scholar]
67. Tipton, KD, Ferrando, AA, Phillips, SM, et al. Postexercise net protein synthesis in human muscle from orally administered amino acids. Am J Physiol. 1999;276(4Pt1):E628–34. PubMed PMID: 10198297. doi: 10.1152/ajpendo.1999.276.4.E628. [PubMed] [CrossRef] [Google Scholar]
68. Rasmussen, BB, Tipton, KD, Miller, SL, et al. An oral essential amino acid-carbohydrate supplement enhances muscle protein anabolism after resistance exercise. J Appl Physiol. 2000;88(2):386–392. PubMed PMID: 10658002. doi: 10.1152/jappl.2000.88.2.386 [PubMed] [CrossRef] [Google Scholar]
69. Waskiw-Ford, M, Hannaian, S, Duncan, J, et al. Leucine-enriched essential amino acids improve recovery from post-exercise muscle damage independent of increases in integrated myofibrillar protein synthesis in young men. Nutrients. 2020;12(4). Epub 2020/04/16. PubMed PMID: 32290521; PMCID: PMC7231404. doi: 10.3390/nu12041061 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
70. Moberg, M, Apro, W, Ekblom, B, et al. Activation of mTORC1 by leucine is potentiated by branched-chain amino acids and even more so by essential amino acids following resistance exercise. Am J Physiol Cell Physiol. 2016;310(11):C874–84. Epub 2016/04/08. PubMed PMID: 27053525. doi: 10.1152/ajpcell.00374.2015 [PubMed] [CrossRef] [Google Scholar]
71. Hannaian, SJ, Hodson, N, Abou Sawan, S, et al. Leucine-enriched amino acids maintain peripheral mTOR-Rheb localization independent of myofibrillar protein synthesis and mTORC1 signaling postexercise. J Appl Physiol. 2020;129(1):133–143. Epub 2020/06/12. PubMed PMID: 32525432; PMCID: PMC7469228. doi: 10.1152/japplphysiol.00241.2020 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
72. Lees, MJ, Wilson, OJ, Webb, EK, et al. Novel essential amino acid supplements following resistance exercise induce aminoacidemia and enhance anabolic signaling irrespective of age: a proof-of-concept trial. Nutrients. 2020;12(7). Epub 2020/07/16. PubMed PMID: 32664648; PMCID: PMC7400893. doi: 10.3390/nu12072067 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
74. Tipton, KD, Rasmussen, BB, Miller, SL, et al. Timing of amino acid-carbohydrate ingestion alters anabolic response of muscle to resistance exercise. Am J Physiol. 2001;281(2):E197–206. PubMed PMID: 11440894. doi: 10.1152/ajpendo.2001.281.2.E197 [PubMed] [CrossRef] [Google Scholar]
75. Fujita, S, Dreyer, HC, Drummond, MJ, et al. Essential amino acid and carbohydrate ingestion before resistance exercise does not enhance postexercise muscle protein synthesis. J Appl Physiol. 2009;106(5):1730–1739. Epub 2017/06/10. PubMed PMID: 18535123; PMCID: PMC2681328. doi: 10.1152/japplphysiol.90395.2008 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
76. Borsheim, E, Tipton, KD, Wolf, SE, et al. Essential amino acids and muscle protein recovery from resistance exercise. Am J Physiol. 2002;283(4):E648–57. PubMed PMID: 12217881. doi: 10.1152/ajpendo.00466.2001 [PubMed] [CrossRef] [Google Scholar]
77. Pasiakos, SM, McClung, HL, Margolis, LM, et al. Human muscle protein synthetic responses during weight-bearing and non-weight-bearing exercise: a comparative study of exercise modes and recovery nutrition. Plos One. 2015;10(10):e0140863. PubMed PMID: 26474292; PMCID: PMC4608805. doi: 10.1371/journal.pone.0140863. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
78. Markofski, MM, Jennings, K, Timmerman, KL, et al. Effect of aerobic exercise training and essential amino acid supplementation for 24 weeks on physical function, body composition, and muscle metabolism in healthy, independent older adults: a randomized clinical trial. J Gerontol A Biol Sci Med Sci. 2019;74(10):1598–1604. Epub 2018/05/12. PubMed PMID: 29750251; PMCID: PMC6748753. doi: 10.1093/gerona/gly109 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
79. Callahan, MJ, Parr, EB, Hawley, JA, et al. Can high-intensity interval training promote skeletal muscle anabolism? Sports Med (Auckland, NZ). 2021;51(3):405–421. Epub 2017/10/17. PubMed PMID: 33512698. doi: 10.1007/s40279-020-01397-3. [PubMed] [CrossRef] [Google Scholar]
80. Forbes, SC, Candow, DG, Smith-Ryan, AE, et al. Supplements and nutritional interventions to augment high-intensity interval training physiological and performance adaptations—A narrative review. Nutrients. 2020;12(2):390. Epub 2020/02/07. PubMed PMID: 32024038; PMCID: PMC7071320. doi: 10.3390/nu12020390. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
81. Hirsch, KR, Greenwalt, CE, Saylor, HE, et al. High-intensity interval training and essential amino acid supplementation: effects on muscle characteristics and whole-body protein turnover. Physiol Rep. 2021;9(1):e14655. Epub 2020/12/29. PubMed PMID: 33369879; PMCID: PMC7769174. doi: 10.14814/phy2.14655 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
82. Hirsch, KR, Greenwalt, CE, Cabre, HE, et al. Metabolic effects of high-intensity interval training and essential amino acids. Eur J Appl Physiol. 2021;121(12):3297–3311. Epub 2020/12/29. PubMed PMID: 34427732. doi: 10.1007/s00421-021-04792-4 [PubMed] [CrossRef] [Google Scholar]
83. Hirsch, KR, Cabre, HE, Gould, LM, et al. Effects of essential amino acids on high-intensity interval training performance, fatigue outcomes, and workload progression. J Am Nutr Assoc. 2023;42(4):411–417. Epub 2020/12/29. PubMed PMID: 35512775. doi: 10.1080/07315724.2022.2060373 [PubMed] [CrossRef] [Google Scholar]
84. Sheffield-Moore, M, Yeckel, CW, Volpi, E, et al. Postexercise protein metabolism in older and younger men following moderate-intensity aerobic exercise. Am J Physiol. 2004;287(3):E513–22. Epub 2004/05/20. PubMed PMID: 15149953. doi: 10.1152/ajpendo.00334.2003 [PubMed] [CrossRef] [Google Scholar]
85. Aquilani, R, D’Antona, G, Baiardi, P, et al. Essential amino acids and exercise tolerance in elderly muscle-depleted subjects with chronic diseases: a rehabilitation without rehabilitation? Biomed Res Int. 2014;2014:341603. Epub 2014/07/11. PubMed PMID: 25009815; PMCID: PMC4070286. doi: 10.1155/2014/341603 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
86. Aquilani, R, Zuccarelli, GC, Dioguardi, FS, et al. Effects of oral amino acid supplementation on long-term-care-acquired infections in elderly patients. Arch Gerontol Geriatr. 2011;52(3):e123–8. Epub 2010/10/12. PubMed PMID: 20934757. doi: 10.1016/j.archger.2010.09.005 [PubMed] [CrossRef] [Google Scholar]
87. Aquilani, R, Viglio, S, Iadarola, P, et al. Oral amino acid supplements improve exercise capacities in elderly patients with chronic heart failure. Am J Cardiol. 2008;101(11A):104E–110E. Epub 2008/07/02. PubMed PMID: 18514618. doi: 10.1016/j.amjcard.2008.03.008 [PubMed] [CrossRef] [Google Scholar]
88. Scognamiglio, R, Piccolotto, R, Negut, C, et al. Oral amino acids in elderly subjects: effect on myocardial function and walking capacity. Gerontology. 2005;51(5):302–308. Epub 2005/08/20. PubMed PMID: 16110231. doi: 10.1159/000086366 [PubMed] [CrossRef] [Google Scholar]
89. Aquilani, R, Zuccarelli, GC, Condino, AM, et al. Despite inflammation, supplemented essential amino acids may improve circulating levels of albumin and haemoglobin in patients after hip fractures. Nutrients. 2017;9(6). Epub 2017/06/22. PubMed PMID: 28635634; PMCID: PMC5490616. doi: 10.3390/nu9060637 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
90. Aquilani, R, Zuccarelli Ginetto, C, Rutili, C, et al. Supplemented amino acids may enhance the walking recovery of elderly subjects after hip fracture surgery. Aging Clin Exp Res. 2019;31(1):157–160. Epub 2018/04/19. PubMed PMID: 29667153. doi: 10.1007/s40520-018-0941-x [PubMed] [CrossRef] [Google Scholar]
91. Baldissarro, E, Aquilani, R, Boschi, F, et al. The hip functional retrieval after elective surgery May be enhanced by supplemented essential amino acids. Biomed Res Int. 2016;2016:9318329. Epub 2016/04/26. PubMed PMID: 27110573; PMCID: PMC4823478. doi: 10.1155/2016/9318329 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
92. Ferrando, A, Bamman, MM, Schutzler, S, et al. Increased nitrogen intake following hip arthroplasty expedites muscle strength recovery. J Aging: Res And Clin Pract. 2013;2(4):369. [Google Scholar]
93. Aquilani, R, Boselli, M, D’Antona, G, et al. Unaffected arm muscle hypercatabolism in dysphagic subacute stroke patients: the effects of essential amino acid supplementation. Biomed Res Int. 2014;2014:964365. Epub 2016/04/26. PubMed PMID: 25431770; PMCID: PMC4241696. doi: 10.1155/2014/964365 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
94. Aquilani, R, Emilio, B, Dossena, M, et al. Correlation of deglutition in subacute ischemic stroke patients with peripheral blood adaptive immunity: essential amino acid improvement. Int J Immunopathol Pharmacol. 2015;28(4):576–583. Epub 2015/10/07. PubMed PMID: 26437899. doi: 10.1177/0394632015608249 [PubMed] [CrossRef] [Google Scholar]
95. Brooks, N, Cloutier, GJ, Cadena, SM, et al. Resistance training and timed essential amino acids protect against the loss of muscle mass and strength during 28 days of bed rest and energy deficit. J Appl Physiol. 2008;105(1):241–248. Epub 2008/05/17. PubMed PMID: 18483167; PMCID: PMC2494840. doi: 10.1152/japplphysiol.01346.2007 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
96. Cholewa, JM, Dardevet, D, Lima-Soares, F, et al. Dietary proteins and amino acids in the control of the muscle mass during immobilization and aging: role of the MPS response. Amino Acids. 2017;49(5):811–820. Epub 2017/02/09. PubMed PMID: 28175999. doi: 10.1007/s00726-017-2390-9 [PubMed] [CrossRef] [Google Scholar]
97. Holloway, TM, McGlory, C, McKellar, S, et al. A novel amino acid composition ameliorates short-term muscle disuse atrophy in healthy young men. Front Nutr. 2019;6:105. Epub 2019/07/30. PubMed PMID: 31355205; PMCID: PMC6636393. doi: 10.3389/fnut.2019.00105 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
98. Killewich, LA, Tuvdendorj, D, Bahadorani, J, et al. Amino acids stimulate leg muscle protein synthesis in peripheral arterial disease. J Vasc Surg. 2007;45(3):554–559. discussion 9-60. Epub 2007/02/27. PubMed PMID: 17321342. doi: 10.1016/j.jvs.2006.11.033 [PubMed] [CrossRef] [Google Scholar]
99. Bauerdick, H, Spellerberg, P, Lamberts, B. Therapy with essential amino acids and their nitrogen-free analogues in severe renal failure. Am J Clin Nutr. 1978;31(10):1793–1796. Epub 1978/10/01. PubMed PMID: 707334. doi: 10.1093/ajcn/31.10.1793 [PubMed] [CrossRef] [Google Scholar]
101. Ferrando, AA, Raj, D, Wolfe, RR. Amino acid control of muscle protein turnover in renal disease. J Ren Nutr. 2005;15(1):34–38. PubMed PMID: 15648004. doi: 10.1053/j.jrn.2004.09.014. [PubMed] [CrossRef] [Google Scholar]
102. Hecking, E, Kohler, H, Zobel, R, et al. Treatment with essential amino acids in patients on chronic hemodialysis: a double blind cross-over study. Am J Clin Nutr. 1978;31(10):1821–1826. Epub 1978/10/01. PubMed PMID: 360819. doi: 10.1093/ajcn/31.10.1821 [PubMed] [CrossRef] [Google Scholar]
103. Unverdi, S, Ceri, M, Uz, E, et al. The effectiveness of oral essential aminoacids and aminoacids containing dialysate in peritoneal dialysis. Ren Fail. 2014;36(9):1416–1419. Epub 2014/09/24. PubMed PMID: 25246343. doi: 10.3109/0886022X.2014.950933 [PubMed] [CrossRef] [Google Scholar]
104. Kato, H, Miura, K, Nakano, S, et al. Leucine-enriched essential amino acids attenuate inflammation in rat muscle and enhance muscle repair after eccentric contraction. Amino Acids. 2016;48(9):2145–2155. Epub 2016/05/12. PubMed PMID: 27168073; PMCID: PMC4989025. doi: 10.1007/s00726-016-2240-1 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
105. Rittig, N, Bach, E, Thomsen, HH, et al. Amino acid supplementation is anabolic during the acute phase of endotoxin-induced inflammation: a human randomized crossover trial. Clin Nutr. 2016;35(2):322–330. Epub 2015/04/22. PubMed PMID: 25896101. doi: 10.1016/j.clnu.2015.03.021. [PubMed] [CrossRef] [Google Scholar]
106. Jones, C, Eddleston, J, McCairn, A, et al. Improving rehabilitation after critical illness through outpatient physiotherapy classes and essential amino acid supplement: a randomized controlled trial. J Crit Care. 2015;30(5):901–907. Epub 2016/05/12. PubMed PMID: 26004031. doi: 10.1016/j.jcrc.2015.05.002 [PubMed] [CrossRef] [Google Scholar]
107. Engelen, M, Safar, AM, Bartter, T, et al. High anabolic potential of essential amino acid mixtures in advanced nonsmall cell lung cancer. Ann Oncol. 2015;26(9):1960–1966. Epub 2015/06/27. PubMed PMID: 26113648; PMCID: PMC4551158. doi: 10.1093/annonc/mdv271 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
109. Dal Negro, RW, Aquilani, R, Bertacco, S, et al. Comprehensive effects of supplemented essential amino acids in patients with severe COPD and sarcopenia. Monaldi Arch Chest Dis. 2010;73(1):25–33. Epub 2010/05/27. PubMed PMID: 20499791. doi: 10.4081/monaldi.2010.310 [PubMed] [CrossRef] [Google Scholar]
110. Dal Negro, RW, Testa, A, Aquilani, R, et al. Essential amino acid supplementation in patients with severe COPD: a step towards home rehabilitation. Monaldi Arch Chest Dis. 2012;77(2):67–75. Epub 2012/12/01. PubMed PMID: 23193843. doi: 10.4081/monaldi.2012.154 [PubMed] [CrossRef] [Google Scholar]
111. Jonker, R, Deutz, NE, Erbland, ML, et al. Effectiveness of essential amino acid supplementation in stimulating whole body net protein anabolism is comparable between COPD patients and healthy older adults. Metabolism. 2017;69:120–129. Epub 2017/03/14. PubMed PMID: 28285641; PMCID: PMC5351771. doi: 10.1016/j.metabol.2016.12.010 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
112. Corsetti, G, Romano, C, Pasini, E, et al. Diet enrichment with a specific essential free amino acid mixture improves healing of undressed wounds in aged rats. Exp Gerontol. 2017;96:138–145. Epub 2017/07/04. PubMed PMID: 28669821. doi: 10.1016/j.exger.2017.06.020 [PubMed] [CrossRef] [Google Scholar]
113. Borsheim, E, Bui, QU, Wolfe, RR. Plasma amino acid concentrations during late rehabilitation in patients with traumatic brain injury. Archives of physical medicine and rehabilitation. Arch Phys Med Rehabil. 2007;88(2):234–238. Epub 2007/02/03. PubMed PMID: 17270522. doi: 10.1016/j.apmr.2006.11.003. [PubMed] [CrossRef] [Google Scholar]
114. Boselli, M, Aquilani, R, Baiardi, P, et al. Supplementation of essential amino acids may reduce the occurrence of infections in rehabilitation patients with brain injury. Nutr Clin Pract. 2012;27(1):99–113. Epub 2012/02/07. PubMed PMID: 22307494. doi: 10.1177/0884533611431068 [PubMed] [CrossRef] [Google Scholar]
115. Borsheim, E, Bui, QU, Tissier, S, et al. Amino acid supplementation decreases plasma and liver triacylglycerols in elderly. Nutrition. 2009;25:281–288. PubMed PMID: 19041223. doi: 10.1016/j.nut.2008.09.001. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
116. Coker, RH, Deutz, NE, Schutzler, S, et al. Nutritional supplementation with essential amino acids and phytosterols may reduce risk for metabolic syndrome and cardiovascular disease in overweight individuals with Mild Hyperlipidemia. J Endocrinol Diabetes Obes. 2015;3(2):Epub 2016/01/05. PubMed PMID: 26726312; PMCID: PMC4696774. [PMC free article] [PubMed] [Google Scholar]
117. Marquis, BJ, Hurren, NM, Carvalho, E, et al. Skeletal muscle acute and chronic metabolic response to essential amino acid supplementation in hypertriglyceridemic older adults. Curr Dev Nutr. 2017;1(11):e002071. Epub 2018/06/30. PubMed PMID: 29955688; PMCID: PMC5998789. doi: 10.3945/cdn.117.002071 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
118. Coker, RH, Miller, S, Schutzler, S, et al. Whey protein and essential amino acids promote the reduction of adipose tissue and increased muscle protein synthesis during caloric restriction-induced weight loss in elderly, obese individuals. Nutr J. 2012;11:105. Epub 2018/06/30. PubMed PMID: 23231757; PMCID: PMC3546025. doi: 10.1186/1475-2891-11-105 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
119. Coker, RH, Shin, K, Scholten, K, et al. Essential amino acid-enriched meal replacement promotes superior net protein balance in older, overweight adults. Clin Nutr. 2019;38(6):2821–2826. Epub 2015/04/22. PubMed PMID: 30638738; PMCID: PMC6588419. doi: 10.1016/j.clnu.2018.12.013. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
121. Jegatheesan, P, Beutheu, S, Ventura, G, et al. Effect of specific amino acids on hepatic lipid metabolism in fructose-induced non-alcoholic fatty liver disease. Clin Nutr. 2016;35(1):175–182. Epub 2015/03/05. PubMed PMID: 25736031. doi: 10.1016/j.clnu.2015.01.021 [PubMed] [CrossRef] [Google Scholar]
122. Theytaz, F, Noguchi, Y, Egli, L, et al. Effects of supplementation with essential amino acids on intrahepatic lipid concentrations during fructose overfeeding in humans. Am J Clin Nutr. 2012;96(5):1008–1016. Epub 2012/10/05. PubMed PMID: 23034968. doi: 10.3945/ajcn.112.035139 [PubMed] [CrossRef] [Google Scholar]
123. Brocca, L, D’Antona, G, Bachi, A, et al. Amino acid supplements improve native antioxidant enzyme expression in the skeletal muscle of diabetic mice. Am J Cardiol. 2008;101(11A):57E–62E. Epub 2008/07/02. PubMed PMID: 18514628. doi: 10.1016/j.amjcard.2008.03.002 [PubMed] [CrossRef] [Google Scholar]
124. Natarajan Sulochana, K, Lakshmi, S, Punitham, R, et al. Effect of oral supplementation of free amino acids in type 2 diabetic patients– a pilot clinical trial. Med Sci Monit. 2002;8(3): CR131–7. Epub 2002/03/12. PubMed PMID: 11887024. [PubMed] [Google Scholar]
125. Pellegrino, MA, Patrini, C, Pasini, E, et al. Amino acid supplementation counteracts metabolic and functional damage in the diabetic rat heart. Am J Cardiol. 2008;101(11A):49E–56E. Epub 2008/07/02. PubMed PMID: 18514627. doi: 10.1016/j.amjcard.2008.03.001 [PubMed] [CrossRef] [Google Scholar]
126. Solerte, SB, Fioravanti, M, Locatelli, E, et al. Improvement of blood glucose control and insulin sensitivity during a long-term (60 weeks) randomized study with amino acid dietary supplements in elderly subjects with type 2 diabetes mellitus. Am J Cardiol. 2008;101(11A):82E–88E. Epub 2008/07/02. PubMed PMID: 18514633. doi: 10.1016/j.amjcard.2008.03.006 [PubMed] [CrossRef] [Google Scholar]
127. Solerte, SB, Gazzaruso, C, Schifino, N, et al. Metabolic effects of orally administered amino acid mixture in elderly subjects with poorly controlled type 2 diabetes mellitus. Am J Cardiol. 2004;93(8A):23A–29A. Epub 2008/07/02. PubMed PMID: 15094102. doi: 10.1016/j.amjcard.2003.11.006 [PubMed] [CrossRef] [Google Scholar]
Nella 4° parte abbiamo analizzato le caratteristiche e funzioni biochimiche dei BCAA. In questa quinta parte, invece, andremo ad analizzare il metabolita della Leucina, l’acido β-idrossi-β-metilbutirrico/HMB.
HMB – storia, ricerca e caratteristiche biochimiche:
L’HMB, o acido β-idrossi-β-metilbutirrico, è un metabolita naturale dell’aminoacido leucina, dove la leucina si converte nel suo analogo cheto-isocaproato (cheto-isocaproato o KIC) e poi si converte in HMB (attraverso l’enzima citosolico KIC diossigenasi[1]);[2] va notato che la versione mitocondriale della KIC diossigenasi converte il KIC nel derivato CoA dell’acido isovalerico (β-idrossiisovalerato).[1]
Tutto l’HMB endogeno deriva dalla leucina[2] e la produzione di HMB è correlata all’assunzione di leucina con la dieta (sembra una cinetica di primo ordine per la KIC diossigenasi citosolica[3][1]), con circa il 5% di tutta l’ossidazione della leucina in vivo che si traduce nella formazione di HMB.[2] Sebbene l’HMB plasmatico tenda a circolare intorno a 1-4µM, può aumentare di 5-10 volte dopo un pasto ricco di leucina.[3]
L’HMB è un metabolita della leucina alimentare nel corpo umano e media una serie di effetti della leucina. L’assunzione di leucina con la dieta può aumentare la formazione di HMB e circa il 5% della leucina alimentare viene convertita in HMB nell’organismo.
Acido β-idrossibutirrico
L’HMB è un membro della famiglia dei composti organici dell’acido carbossilico.[4] È un analogo strutturale dell’acido butirrico con un gruppo funzionale idrossile e un sostituente metile situato sul carbonio beta.[4][5] Per estensione, altri analoghi strutturali includono l’acido β-idrossibutirrico e l’acido β-metilbutirrico.[4][5]
Aleksander Mikhaylovich Zaytsev
La prima sintesi chimica dell’HMB è stata pubblicata nel 1877 dai chimici russi Michael e Alexander Zaytsev.[6] L’HMB è stato isolato dalla corteccia dell’Erythrophleum couminga (un albero del Madagascar) nel 1941 da Leopold Ružička.[7] L’isolamento più precoce dell’HMB come metabolita umano è stato effettuato da Tanaka e collaboratori nel 1968 da un paziente affetto da acidemia isovalerica.[8][9]
Gli effetti dell’HMB sul muscolo scheletrico umano sono stati scoperti per la prima volta da Steven L. Nissen della Iowa State University a metà degli anni ’90.[8][10] Nissen ha fondato un’azienda chiamata Metabolic Technologies, Inc. (MTI) all’epoca della sua scoperta, che in seguito ha acquisito sei brevetti relativi all’HMB che l’azienda ha utilizzato per concedere in licenza il diritto di produrre e incorporare l’HMB negli integratori alimentari. [10][11][12] Quando è stato commercializzato per la prima volta alla fine degli anni ’90, l’HMB è stato commercializzato esclusivamente come integratore per l’esercizio fisico, per aiutare gli atleti e i bodybuilder a costruire i muscoli.[11] MTI ha successivamente sviluppato due prodotti contenenti HMB, Juven e Revigor, di cui Abbott Nutrition ha ottenuto i diritti di commercializzazione rispettivamente nel 2003 e nel 2008.[8][11] Da allora, Abbott ha commercializzato Juven come alimento medico e il marchio Revigor di HMB come ingrediente attivo in prodotti alimentari (ad es, alcune formulazioni di Ensure) e altri alimenti medici (ad esempio, alcune formulazioni di Juven).[8][13][11]
Sono state sviluppate diverse vie sintetiche per l’HMB. Le prime sintesi chimiche riportate hanno avvicinato l’HMB all’ossidazione di precursori alchenici, dioli vicinali e alcol:
nel 1877, i chimici russi Michael e Alexander Zaytsev riportarono la preparazione dell’HMB per ossidazione del 2-metilpent-4-en-2-olo con acido cromico (H2CrO4);[6]
nel 1880 e nel 1889, Schirokoff e Reformatsky (rispettivamente) riportarono che la scissione ossidativa del diolo vicinale 4-metilpentano-1,2.,4-triolo con potassio acidificato, 4-triolo con permanganato di potassio acidificato (KMnO4) produce HMB[14][15] – questo risultato è più vicino alla prima sintesi, poiché il KMnO4 diluito a freddo ossida gli alcheni a cis-dioli vicinali che il KMnO4 acido a caldo ossida ulteriormente a composti contenenti carbonile, mentre l’intermedio diolo non si ottiene quando si utilizzano condizioni acide a caldo per l’ossidazione degli alcheni. [In altre parole, il 4-metilpentano-1,2,4-triolo racemico è un derivato del 2-metilpent-4-en-2-olo e l’acido β-idrossi-β-metilbutirrico è un derivato di entrambi,
nel 1892, Kondakow riportò la preparazione dell’HMB per ossidazione con permanganato del 3-metilbutano-1,3-diolo.
A seconda delle condizioni sperimentali, la cicloaddizione di acetone e chetene produce il β-isovalerolattone o il 4,4-dimetilossetan-2-one,[16][17] che si idrolizzano entrambi in condizioni basiche per produrre la base coniugata dell’HMB. La reazione aloformica fornisce un’altra via per l’HMB che comporta l’alogenazione esaustiva della regione metil-chetonica dell’alcol di diacetone con ipobromito di sodio o ipoclorito di sodio;[5][18][19] l’alcol di diacetone è facilmente disponibile dalla condensazione aldolica dell’acetone. [Un approccio organometallico all’HMB prevede la carbossilazione dell’alcol tert-butilico con monossido di carbonio e reagente di Fenton (perossido di idrogeno e ferro).[5][20] In alternativa, l’HMB può essere preparato attraverso l’ossidazione microbica dell’acido β-metilbutirrico da parte del fungo Galactomyces reessii.[21]
HMB nella supplementazione sportiva:
Una formulazione di HMB disponibile in commercio. Ogni capsula di gelatina formato 000 contiene 1 grammo di HMB-Ca e una quantità non specificata di cellulosa microcristallina e magnesio stearato.
L’HMB può essere integrato sotto forma di sale di calcio monoidrato (comunemente chiamato HMB di calcio) o come acido libero, ovvero HMB senza il sale di calcio. Il sale di calcio ha una costante di dissociazione simile a quella dell’acetato di calcio[22] e ha un Tmax dell’ordine di 1-2 ore dopo l’ingestione di 1 g di Ca-HMB, con un picco di 487,9+/-19,0nmol/mL (Cmax) e un’emivita di 2,5 ore. Uno studio successivo, condotto con 1 g di HMB calcico, ha rilevato una Cmax di 131+/-10µmol/L e un ritorno al valore basale dopo 12 ore;[23] il motivo di questa discrepanza con la stessa dose non è noto.
Confrontando l’acido libero con il sale di calcio (livelli equivalenti di HMB, quindi 0,8 g di acido libero contro 1 g di HMB di calcio), la Cmax è più alta con l’acido libero del 76-97% e il Tmax più breve (30 minuti), mentre anche l’AUC è aumentata del 91-97%.[23] Quando si tiene la dose di acido libero per via sublinguale per 15 minuti prima di deglutire, non sembrano esserci differenze significative rispetto alla semplice deglutizione.[23]
La forma di acido libero sembra essere assorbita meglio e raggiungere il picco sierico più rapidamente rispetto alla forma di sale di calcio dell’HMB.
Di solito, quando si parla di integrazione alimentare negli atleti, si utilizza una dose di 3 g di HMB. Ciò è dovuto principalmente al fatto che si tratta della dose più comunemente utilizzata, ma le prove limitate che confrontano 3 g con dosi più elevate (di solito 6 g) non trovano alcuna differenza significativa tra le due dosi.[24]
6 g di HMB non sembrano essere significativamente migliori di 3 g di HMB.
Massa muscolare:
Per quanto riguarda gli studi sugli animali, 460mg/kg di HMB al giorno somministrati a ratti di mezza età sembrano essere efficaci nel ridurre il tasso di declino motorio e l’area della sezione trasversale muscolare durante il successivo processo di invecchiamento, ma non sono riusciti a influenzare la massa magra.[25] Quando questa dose viene somministrata a ratti di sesso femminile di età avanzata, l’aumento della massa muscolare e della produzione di potenza osservato con l’esercizio fisico non viene incrementato.[26]
Gli studi sull’uomo sono in qualche modo simili, con 2 g di HMB (integratore combinato con 5 g di L-arginina e 1,5 g di L-lisina) in grado di migliorare il controllo muscolare e la potenza in uscita per 12 settimane in donne (età media 76 anni. 7) senza influire sulla massa magra[27], anche se il primo studio ha rilevato una tendenza all’aumento della massa magra (e i test in acuto hanno evidenziato un aumento del 20% della sintesi proteica[27]), mentre uno studio successivo ha confermato un aumento della massa magra, ma senza miglioramenti della funzione muscolare.[28] Uno studio con l’aggiunta di vitamina D ha riscontrato benefici sia sulla forza che sulla massa magra nel corso di un anno.[29]
Schema delle cascate di segnalazione biomolecolare anabolica coinvolte nella sintesi proteica del muscolo miofibrillare e nella biogenesi mitocondriale in risposta all’esercizio fisico e a specifici aminoacidi o loro derivati (principalmente l-leucina e HMB).
Negli adulti anziani che partecipano all’allenamento con i pesi, l’HMB supplementare è associato a un aumento della massa magra (0,8 kg in 8 settimane) senza influire sulla massa grassa.[30]
È possibile che l’integrazione di HMB nella dieta degli anziani attenui il tasso di perdita muscolare che si verifica durante il processo di invecchiamento.
L’HMB possiede proprietà mitogeniche, valutate da cellule muscolari umane quiscienti stimolate a proliferare con l’incubazione dell’HMB, con un picco di efficacia (aumento della MyoD) a 50ug/mL in questo studio ed effetti negativi a 200ug/mL.[31] Questo effetto mitogenico diretto è stato notato altrove,[32][33] e suggerisce che l’HMB può indurre le cellule muscolari quiscienti (dormienti) alla differenziazione cellulare.
Con l’integrazione di HMB è stata notata una proliferazione cellulare secondaria alla via MAPK/ERK, poiché gli inibitori di MEK aboliscono gli effetti proliferativi dell’HMB in vitro.[31] Questa via è nota per essere un regolatore della proliferazione delle cellule muscolari[34][35] e sembra mediare la proliferazione cellulare indotta dall’HMB.[31]
La via dell’MAPK
L’HMB può indurre la proliferazione delle cellule muscolari attraverso la via MAPK/ERK, che è uno dei bersagli molecolari dell’integrazione di HMB.
Esaminando le vie molecolari, è stato riscontrato che l’HMB stimola la sintesi proteica muscolare attraverso la via mTOR[36] a valle di PI3K/Akt[31] e può avvenire indipendentemente dalla leucina.[37][31] Nei ratti (320mg/kg) è stato riscontrato un aumento dell’espressione di mTOR (429,2%) e la successiva fosforilazione di p70S6K.[36]
È stato osservato che gli inibitori di Akt inibiscono la differenziazione muscolare indotta dall’HMB (suggerendo che sia fondamentale per la segnalazione)[31] ed è stato ipotizzato che la via di segnalazione di Akt medi la differenziazione delle cellule muscolari[31].
La sintesi proteica muscolare sembra essere mediata dalla via mTOR (a valle della segnalazione di Akt, il secondo bersaglio molecolare dell’HMB) e dalla successiva fosforilazione di p70S6K.
L’HMB è coinvolto nella riduzione dell’apoptosi (morte cellulare regolata) dei miociti e delle cellule satelliti e, grazie a questi effetti anti-apoptotici, si pensa che l’integrazione di HMB possa svolgere un ruolo in situazioni caratterizzate dall’apoptosi dei miociti (catabolismo associato all’invecchiamento,[38][39] distrofie muscolari,[40][41] e cachessia[42][43]). È stato confermato in vitro che l’HMB riduce l’apoptosi aumentando il rapporto tra Bcl-2/Bcl-X e Bax[31-18] attraverso la segnalazione di Akt[44] che porta le proteine antiapoptotiche Bcl-2 e Bcl-X a sequestrare le proteine pro-apoptotiche Bax.[45]
Analogamente all’induzione della sintesi proteica e della differenziazione muscolare, gli effetti anti-apoptotici dell’HMB sono a valle della segnalazione di Akt.
Danno Muscolare:
LDH
L’integrazione di 3 g di HMB (l’uso del sale di calcio o dell’acido libero non è stato rivelato) prima dell’esercizio fisico in maschi non allenati non ha alterato in modo significativo i livelli di creatinchinasi, sebbene l’integrazione prima dell’esercizio sembrasse ridurre l’LDH sierico.[46] Uno studio successivo, che ha replicato i risultati ma ha utilizzato una forma di sale libero di HMB (assorbito più velocemente[23]), ha osservato che la creatinchinasi indotta dall’esercizio fisico in maschi allenati è stata ridotta (dal 329% al 104%) dopo 3 g di HMB acido libero.[47]
Negli studi che valutano l’indolenzimento muscolare, 3 g di HMB prima dell’esercizio in uomini non allenati non hanno ridotto l’indolenzimento[46], anche se 3 g (di acido libero piuttosto che di sale di calcio) prima dell’esercizio hanno migliorato la capacità percepita degli atleti di eseguire gli allenamenti nei pochi giorni successivi al test.[35] Raddoppiare la dose a 6 g di sale di calcio non ha causato una riduzione dell’indolenzimento acuto.[48]
Sono stati condotti due studi sull’integrazione di HMB e sul recupero. Entrambi hanno utilizzato l’HMB alla dose di 3 g di sale di calcio (con 0,3 g di CCI) e uno ha rilevato che l’integrazione ha favorito il recupero dal sollevamento pesi quando è stata misurata nei tre giorni successivi all’esercizio[49], mentre l’altro studio, che ha utilizzato la stessa dose per favorire il recupero dalla corsa in discesa, non ha riscontrato benefici;[50] quest’ultimo studio, tuttavia, potrebbe aver utilizzato un integratore privo di HMB[51], il che potrebbe spiegare il fallimento.
Non è chiaro se l’integrazione di HMB sia in grado di ridurre l’indolenzimento muscolare, con prove limitate che valutano i tassi di recupero e che suggeriscono che sia l’HMB acido libero sia l’HMB sale di calcio possano avere dei benefici.
Sintesi Proteica Muscolare:
Uno studio che ha confrontato gli effetti di 3,42 g di HMB con la stessa dose orale di leucina ha rilevato che mentre l’HMB ha aumentato la sintesi proteica muscolare (valutata mediante traccianti di fenilalanina incorporati nei miociti) del 70%, la leucina ha aumentato la sintesi proteica muscolare del 110%.[52]
Sembra essere meno efficace di una pari dose orale di leucina nel promuovere la sintesi proteica muscolare.
È stato osservato che l’aggiunta di 3 g di HMB alla dieta di atleti sottoposti ad allenamento fisico aumenta la massa muscolare dello 0,2+/-2,2% nell’arco di 9 settimane, sebbene questo studio sia confuso con un aumento dell’8% dell’assunzione di cibo (e una riduzione del 10% del placebo)[53] e questo studio si scontra con altri due condotti su persone non allenate, in cui si osserva che l’HMB induce la sintesi proteica muscolare sia nei gruppi ad alto (175 g) che a basso (117 g) contenuto proteico[7] e che non vi sono differenze dovute al sesso o allo stato di allenamento. [54] Anche l’unico studio condotto su giovani atleti ha riportato risultati benefici, ma la composizione della dieta non è stata resa nota (solo una dichiarazione che non presentava differenze).[55]
Al contrario, uno studio comparativo tra 3 g di HMB in formulazione a rilascio ritardato e sale di calcio standard non ha riscontrato un effetto per 6 settimane in nessuno dei due gruppi[56] e il raddoppio della dose a 6 g di calcio-HMB (somministrato tramite frullato proteico) non ha superato il placebo (frullato proteico simile senza HMB) per 28 giorni.[57] Sono stati riportati risultati nulli anche in persone non allenate,[58] a sostegno dell’idea che lo stato di allenamento sia irrilevante.
Le prove a sostegno dell’idea che l’integrazione di HMB promuova la sintesi proteica muscolare negli atleti allenati a 3 g al giorno sono scarse e probabilmente non vi è alcun beneficio.
Atrofia Muscolare/Catabolismo:
L’HMB possiede un effetto anticatabolico (preserva la massa muscolare) che si ritiene sia in qualche modo nuovo rispetto all’integrazione di leucina, in quanto gli effetti soppressivi della leucina sulla massa muscolare sono massimi a 5-10mM[59] (nettamente superiori ai livelli a digiuno di 0,1mM[60][61] e alle concentrazioni postprandiali che sono state osservate circa raddoppiate dopo infusioni di 162-261mg/kg/h[62]) nonostante le concentrazioni raggiungibili con l’HMB. 1mM[60-51][61] e delle concentrazioni postprandiali che sono state osservate come circa raddoppiate dopo infusioni di 162-261mg/kg/h[62]), nonostante le concentrazioni raggiungibili con la leucina siano sufficienti a promuovere la sintesi proteica muscolare[63] (in misura maggiore rispetto all’HMB[44]), ma la leucina a 0,5mM sembra avere scarsi effetti anticatabolici (6,7% in questo modello animale che ha osservato un aumento della sintesi del 36-38%[64]). È possibile che l’HMB svolga un ruolo di agente anticatabolico nonostante il suo scarso effetto sulla sintesi proteica muscolare, e ciò è in qualche modo supportato dal fatto che gli effetti anticatabolici della leucina sono 10-20 volte superiori alla concentrazione necessaria per promuovere la sintesi proteica muscolare[59] e che circa il 5% della leucina viene convertito in HMB nell’organismo.[6]
È plausibile che l’HMB sia il metabolita anticatabolico della leucina, mentre da solo non è in grado di superare la leucina nella sintesi proteica muscolare (forse perché altri metaboliti della leucina sono più potenti nell’indurre la sintesi proteica), ma può avere un ruolo nella prevenzione della perdita muscolare che non richiede gli altri metaboliti della leucina né la leucina stessa.
A 50μM, si è notato che l’HMB riduce l’atrogina-1 basale in vitro e l’induzione dell’atrogina-1 da parte di stimoli catabolici,[65] che sembra essere una concentrazione raggiungibile di HMB associata a un aumento della sintesi proteica muscolare. [29][18] Ciò suggerisce che gli effetti anticatabolici dell’HMB sono rilevanti (poiché l’atrogin-1 è una proteina che media la disgregazione delle proteine muscolari[66]) e, sebbene siano in parte a valle della segnalazione di mTOR[29], sono completamente dipendenti dall’attivazione di p38/MAPK (p42/44 MAPK sembra non essere coinvolta).[68][65]
Gli effetti anticatabolici (in vitro) sono stati confermati nei confronti dei glucocorticoidi,[65] degli stimoli proinfiammatori LPS[68][24] e TNF-α,[69][24] e dell’angiotensione II.[69][24]
Le ricerche in vitro supportano l’idea che l’HMB sia anticatabolico, e questo effetto anticatabolico sembra estendersi a un’ampia varietà di fattori di stress catabolico e si verifica a una concentrazione raggiungibile dopo l’ingestione orale di integratori di HMB. Ciò avviene attraverso la segnalazione p38/MAPK
Ciò è stato osservato con 3 g di sali di HMB per 10 giorni in adulti anziani sottoposti a riposo a letto, invertendo il declino della massa magra (2,05+/-0,66 kg) a nessun cambiamento significativo (0,17+/-0,19 kg con tendenza all’aumento);[70] che è simile agli aminoacidi a catena ramificata e alla leucina isolata. [71][72] Altri studi hanno osservato che l’integrazione di HMB è efficace nell’attenuare il tasso di perdita di massa magra osservato nella cachessia da cancro[73][74][30] e una combinazione di HMB con L-arginina e L-glutammina ha mostrato efficacia nei pazienti affetti da AIDS[75], anche se in vitro non sembrano avere un effetto anticatabolico sinergico.[29] Attualmente, gli effetti anticatabolici della leucina e dell’HMB non sono stati confrontati direttamente.
Uno studio in acuto che ha utilizzato 3,42 g di HMB rispetto a 3,42 g di leucina ha osservato che mentre la leucina ha superato l’HMB sulla sintesi proteica muscolare, l’HMB è stato in grado di attenuare la disgregazione delle proteine muscolari (57%).[44]
Gli studi sugli atleti volti a valutare la disgregazione delle proteine muscolari sono limitati; uno studio che ha utilizzato 3 g di HMB come sale di calcio per 3 giorni in atlete di judo d’élite durante una grave restrizione calorica (20 kcal/kg e 1,33 g/kg di proteine; per simulare la situazione prima di una gara) non è riuscito a superare il placebo.[36]
Uno studio condotto su atleti di pallavolo d’élite (giovani) non ha rilevato differenze nel cortisolo dopo l’integrazione di 3 g di HMB per un periodo di 7 settimane in concomitanza con l’allenamento.[46]
È stato confermato che l’integrazione di HMB è anticatabolica nei periodi di deperimento muscolare ad alto rischio (cachessia oncologica, AIDS, degenza a letto) a un dosaggio supplementare fattibile, ma non ci sono prove sufficienti per valutare correttamente il suo ruolo negli atleti. Sembra essere migliore della leucina in questo, ma richiede prove più solide per essere confermata.
Appetito:
Esistono alcuni studi che somministrano HMB a 3 g a maschi allenati alla resistenza che riportano cambiamenti nell’assunzione di cibo, come ad esempio 9 settimane di integrazione che causano una tendenza all’aumento dell’assunzione calorica complessiva e un aumento significativo dell’assunzione di grassi (totali, saturi e monoinsaturi del 44%, 44% e 53% rispetto al basale)[37] e altrove è stato notato che i gruppi integrati con HMB consumano più proteine rispetto al placebo (questo studio ha notato una diminuzione rispetto al basale nel placebo che non era presente nell’HMB); [76] quest’ultimo studio non ha riscontrato differenze nell’assunzione di grassi, ma ha rilevato un aumento relativo dell’apporto calorico. [76]
Altri studi non hanno rilevato differenze significative nella composizione o nella quantità della dieta con 3 g di HMB in gruppi demografici simili[38] e giovani.[77] Alcuni risultati nulli sono stati ottenuti con interventi dietetici (standardizzazione della dieta o introduzione di supplementi calorici, che controllano l’appetito).[78]
Alcuni interventi sull’uomo notano che i gruppi integrati con HMB a 3 g tendono a mangiare di più, anche se questo aumento dell’assunzione di cibo non è affidabile per quanto riguarda la frequenza con cui si verifica e quali macronutrienti vengono consumati in eccesso. Non è certo che l’HMB abbia un ruolo causale in questo caso.
Profilo di sicurezza:
I test tossicologici hanno rilevato che il livello senza effetti avversi osservati (NOAEL; la dose più alta non associata a segni di tossicità) per l’ingestione orale di HMB nei ratti è di 3490mg/kg per i ratti maschi e 4160mg/kg per le femmine;[79] si tratta di un equivalente umano stimato[80-71] di 558mg/kg e 665mg/kg, e ipotizzando un peso corporeo di 150lbs equivale a 38g (maschi) e 45g (femmine). Altri test tossicologici sugli animali includono una dose di circa 5 g/kg nei maiali per 4 giorni, che non ha alterato alcun parametro biochimico o il peso degli organi (Nutritional role of the leucine metabolite B-hydroxy B-methylbutyrate (HMB) 1997; citato tramite una revisione[81]).
Studi tossicologici sull’uomo hanno osservato che circa 6 g di HMB al giorno (78 mg/kg) per un mese in giovani maschi non allenati e sottoposti a esercizio fisico non hanno mostrato effetti tossici sui parametri sierici (metà della dose ha avuto un aumento spontaneo dei basofili, considerato insignificante)[82] e 3 g di HMB al giorno per un massimo di 8 settimane sia in giovani che in anziani non hanno alterato i parametri tossicologici nel siero[83] e questa dose è risultata sicura per un anno di somministrazione (studio confuso con l’ingestione di L-lisina e L-arginina). [Nel complesso, le dosi standard di HMB sembrano essere ben tollerate per lunghi periodi di tempo (meta-analisi).[84]
È stato dimostrato che l’integrazione di HMB fino a 3 g al giorno è molto ben tollerata e si sospetta che dosi maggiori siano altrettanto sicure (ma con meno test sull’uomo). L’integrazione di HMB non desta troppe preoccupazioni in termini di sicurezza.
Conclusioni:
Come abbiamo visto, l’HMB può essere utile in determinate circostanze sebbene il suo margine di efficacia sia tutto sommato sorretto su deboli evidenze scientifiche.
Tralasciando il suo dubbio effetto migliorativo sulla sintesi proteica, sembrerebbe che un suo utilizzo in contesti di ipocalorica e, quindi, tendenzialmente catabolici potrebbe offrire un certo vantaggio. Di conseguenza, il suo uso, se lo si vuole prendere in considerazione, potrebbe essere circoscritto al “Cut” o “Pre-Gara” alla dose di 3-9g/die.
La sua aggiunta in fasi di “Bulk” non ha praticamente mai dimostrato di apportare vantaggi anche minimi rispetto al suo mancato inserimento.
Gabriel Bellizzi [CEO BioGenTech]
Riferimenti:
Coffman DD, Cramer R, Mochel WE (June 1958). “Syntheses by Free-radical Reactions. V. A New Synthesis of Carboxylic Acids”. Journal of the American Chemical Society. 80 (11): 2882–2887. doi:10.1021/ja01544a072.
“3-OH-isovaleric acid”. ChemSpider. Royal Society of Chemistry. 2015. Archived from the original on 11 August 2016. Retrieved 10 August 2016. Experimental Boiling Point: … 128 °C / 7 mm … Experimental solubility: Soluble in water
“beta-Hydroxyisovaleric acid”. PubChem Compound. United States National Library of Medicine – National Center for Biotechnology Information. 3 February 2018. Archived from the original on 6 February 2018. Retrieved 6 February 2018. Chemical Names: Beta-Hydroxyisovaleric acid; 3-Hydroxy-3-methylbutanoic acid; … 3-Hydroxyisovaleric acid; 3-Hydroxy-3-methylbutyric acid
“3-hydroxyisovaleric acid”. Chemical Entities of Biological Interest. European Bioinformatics Institute. 23 October 2015. Archived from the original on 12 March 2016. Retrieved 20 August 2016.
The earliest citation for the synthesis of β-hydroxy β-methylbutyric acid in the Reaxys chemical database as of September 2016 is: Saytzeff M, Saytzeff A (1877). “Synthese des Allyldimethylcarbinols” [Synthesis of allyldimethylcarbinols]. Justus Liebig’s Annalen der Chemie (in German). 185 (2–3): 151–169. doi:10.1002/jlac.18771850204.
Ružička L, Dalma G, Engel BG, Scott WE (1941). “Zur Kenntnis der Erythrophleum-Alkaloide. (5. Mitteilung). Identifizierung der niedermolekularen Spaltsäure des Coumingins” [Concerning erythrophleum alkaloids. (5th Communication). Identification of the low molecular weight cleavage acids from coumingin]. Helvetica Chimica Acta (in German). 24 (1): 1449–1458. doi:10.1002/hlca.194102401171.
Tanaka K, Orr JC, Isselbacher KJ (May 1968). “Identification of beta-hydroxyisovaleric acid in the urine of a patient with isovaleric acidemia”. primary source. Biochimica et Biophysica Acta (BBA) – Lipids and Lipid Metabolism. 152 (3): 638–41. doi:10.1016/0005-2760(68)90107-0. PMID5656832.
Gresham TL, Jansen JE, Shaver FW, Beears WL (January 1954). “β-Propiolactone. XIV. β-Isovalerolactone”. Journal of the American Chemical Society. 76 (2): 486–488. doi:10.1021/ja01631a045.
WO application 2012140276, Noti C, Schmid L, Rittiner B, Hanselmann P, Bierstedt A, “Process for the preparation of 3-hydroxy-3-methylbutyric acid or its calcium salts”, published 10 January 2013, assigned to Lonza Ltd
Jump up to:ab Kohn M (September 1903). “Zur Kenntnis des Diacetonalkohols und des Mesityloxyds” [Knowledge of diacetone alkohols and mesityl oxide]. Monatshefte für Chemie und Verwandte Teile Anderer Wissenschaften. 24 (9): 765–772. doi:10.1007/BF01526057. S2CID96317019.
Doraiswamy LK (February 2001). “Example 5.2”. Organic Synthesis Engineering. New York: Oxford University Press. pp. 102–124. ISBN978-0-19-509689-7.
Nella 3° parte abbiamo analizzato le caratteristiche e funzioni biochimiche della L-Citrullina e della L-Arginina. In questa quarta parte, invece, andremo ad analizzare uno degli integratori a base di AA più discussi in assoluto: gli Aminoacidi a Catena Ramificata/BCAA.
BCAA – caratteristiche biochimiche:
Un aminoacido a catena ramificata (BCAA) è un aminoacido con una catena laterale alifatica con un ramo (un atomo di carbonio centrale legato a tre o più atomi di carbonio). Tra gli aminoacidi proteinogenici, vi sono tre BCAA: leucina, isoleucina e valina.[1] Tra i BCAA non proteinogenici vi sono l’acido 2-aminoisobutirrico e l’alloisoleucina.
I tre BCAA proteinogenici sono tra i nove aminoacidi essenziali per l’uomo, rappresentando il 35% degli aminoacidi essenziali nelle proteine muscolari e il 40% degli aminoacidi preformati richiesti dai mammiferi.[2] La sintesi dei BCAA avviene in tutti i luoghi delle piante, all’interno dei plastidi della cellula, come determinato dalla presenza di mRNA che codificano per gli enzimi della via metabolica.[3][4][5] L’ossidazione dei BCAA può aumentare l’ossidazione degli acidi grassi e svolgere un ruolo nell’obesità. Fisiologicamente, i BCAA svolgono un ruolo nel sistema immunitario e nella funzione cerebrale. I BCAA vengono scomposti efficacemente dagli enzimi deidrogenasi e decarbossilasi espressi dalle cellule immunitarie e sono necessari per la crescita e la proliferazione dei linfociti e per l’attività dei linfociti T citotossici.[4] Infine, i BCAA condividono con gli aminoacidi aromatici (Trp, Tyr e Phe) la stessa proteina di trasporto nel cervello. Una volta nel cervello, i BCAA possono avere un ruolo nella sintesi proteica, nella sintesi dei neurotrasmettitori e nella produzione di energia.[4]
Aspartato transaminasi [aminotrasferasi] da E. coli con cofattore Piridossal 5′ Fosfato
Cinque enzimi partecipano alle vie di sintesi parallele di isoleucina, valina e leucina: la treonina deidrogenasi, l’acetoidrossiacido sintasi, la chetoacido reduttasi, la diidrossiacido deidrogenasi e l’aminotransferasi.[3] La treonina deidrogenasi catalizza la deaminazione e la disidratazione della treonina a 2-chetobutirrato e ammoniaca. L’isoleucina forma un circuito di feedback negativo con la treonina deidrogenasi. L’acetoidrossiacido sintasi è il primo enzima della via parallela che esegue la reazione di condensazione in entrambe le fasi: condensazione del piruvato ad acetolattato nella via della valina e condensazione del piruvato e del 2-chetobutirrato per formare acetoidrossibutirrato nella via dell’isoleucina. Successivamente, la chetoacido-riduttasi riduce gli acetoidrossiacidi della fase precedente per produrre diidrossiacidi in entrambe le vie della valina e dell’isoleucina. La diidrossiacido deidrogenasi converte i diidrossiacidi nella fase successiva. La fase finale della via parallela è condotta dall’amino-transferasi, che produce i prodotti finali di valina e isoleucina.[3] Una serie di altri quattro enzimi – isopropilmalato sintasi, isopropilmalato isomerasi, isopropilmalato deidrogenasi e aminotransferasi – è necessaria per la formazione della leucina dal 2-ossolsovalerato.[3]
La degradazione degli aminoacidi a catena ramificata coinvolge il complesso della deidrogenasi degli alfa-chetoacidi a catena ramificata (BCKDH). Una carenza di questo complesso porta a un accumulo di aminoacidi a catena ramificata (leucina, isoleucina e valina) e dei loro sottoprodotti tossici nel sangue e nelle urine, dando alla condizione il nome di malattia delle urine a sciroppo d’acero. D’altra parte, l’attività incontrollata di questo complesso causa un deficit di chetoacido deidrogenasi a catena ramificata.
La degradazione di leucina, isoleucina e valina. Viene illustrata anche la via di degradazione della metionina.
Il complesso BCKDH converte gli aminoacidi a catena ramificata in derivati dell’acil-CoA, che dopo successive reazioni vengono convertiti in acetil-CoA o succinil-CoA che entrano nel ciclo dell’acido citrico.[6]
Gli enzimi coinvolti sono l’aminotransferasi a catena ramificata e la 3-metil-2-ossobutanoato deidrogenasi.
Mentre la maggior parte degli aminoacidi viene ossidata nel fegato, i BCAA vengono ossidati principalmente nel muscolo scheletrico e in altri tessuti periferici.[4] Sono stati testati gli effetti della somministrazione di BCAA sulla crescita muscolare del diaframma di ratto e si è concluso che non solo una miscela di BCAA da sola ha lo stesso effetto sulla crescita di una miscela completa di aminoacidi, ma che una miscela di aminoacidi con tutti i BCAA tranne i BCAA non influisce sulla crescita muscolare del diaframma di ratto. [7] La somministrazione di isoleucina o valina da sole non ha influenzato la crescita muscolare, anche se la somministrazione di leucina da sola sembra essere quasi altrettanto efficace della miscela completa di BCAA. La leucina attiva indirettamente la p70 S6 chinasi e stimola l’assemblaggio del complesso eIF4F, essenziali per il legame dell’mRNA nell’avvio della traslazione.[7] La p70 S6 chinasi fa parte della via di segnalazione del complesso mammalian target of rapamycin (mTOR) ed è stato dimostrato che consente l’ipertrofia adattiva e il recupero del muscolo di ratto. [8] A riposo, l’infusione di proteine stimola la sintesi proteica 30 minuti dopo l’inizio dell’infusione e la sintesi proteica rimane elevata per altri 90 minuti.[9] L’infusione di leucina a riposo produce un effetto stimolante di sei ore e un aumento della sintesi proteica attraverso la fosforilazione della p70 S6 chinasi nei muscoli scheletrici.[10] Dopo l’esercizio di resistenza, senza la somministrazione di BCAA, una sessione di esercizio di resistenza non influisce sulla fosforilazione di mTOR e produce addirittura una diminuzione della fosforilazione di Akt. È stata rilevata una certa fosforilazione della p70 S6 chinasi. Quando sono stati somministrati BCAA dopo una sessione di allenamento, una fosforilazione sufficiente di p70 S6 chinasi e S6 ha indicato l’attivazione della cascata di segnalazione.[10]
Metabolismo degli aminoacidi a catena ramificata nel muscolo scheletrico. (1) I BCAA sono transaminati con α-chetoglutarato dalla BCAA transaminasi per generare glutammato. (2) La deaminazione del glutammato produce α-chetoglutarato e ammoniaca. (3) Durante l’esercizio fisico, l’AMP viene generato dalla degradazione dell’ATP nel muscolo scheletrico. La reazione dell’AMP deaminasi muscolare forma anche ammoniaca. (4) La glutammina si forma da ammoniaca e glutammato, una reazione catalizzata dalla glutammina sintetasi. (5) L’α-chetoglutarato formato dalla glutammato deidrogenasi può entrare anapleroticamente nel ciclo TCA. Tratto e modificato da Groper e Smith (2013).
Oltre alla segnalazione cellulare, la via mTOR svolge anche un ruolo nella crescita delle cellule beta che porta alla secrezione di insulina.[11] L’elevata presenza di glucosio nel sangue avvia il processo della via di segnalazione mTOR, in cui la leucina svolge un ruolo indiretto.[9][12] La combinazione di glucosio, leucina e altri attivatori fa sì che mTOR inizi a segnalare la proliferazione delle cellule beta e la secrezione di insulina. Concentrazioni più elevate di leucina causano un’iperattività della via mTOR e l’attivazione della chinasi S6 porta all’inibizione del substrato del recettore dell’insulina attraverso la fosforilazione della serina.[11][12] Nella cellula l’aumento dell’attività del complesso mTOR causa l’eventuale incapacità delle cellule beta di rilasciare insulina e l’effetto inibitorio della chinasi S6 porta all’insulino-resistenza nelle cellule, contribuendo allo sviluppo del diabete di tipo 2.[12]
La metformina è in grado di attivare l’AMP chinasi che fosforila le proteine coinvolte nella via mTOR e porta alla progressione del complesso mTOR dallo stato inattivo a quello attivo.[12] Si suggerisce che la Metformina agisca come inibitore competitivo dell’amminoacido leucina nella via mTOR.
BCAA e Sport:
Sappiamo che la Leucina, in particolare, attiva mTOR, un segnale anabolico che media la sintesi proteica muscolare [13], a sua volta correlata agli adattamenti della forza e dell’ipertrofia [14]. A tal fine, si è ipotizzato che i BCAA siano utili per le prestazioni, il recupero e la composizione corporea [15].
Una recente pubblicazione sostiene che l’assunzione di livelli accettabili di BCAA dovrebbe essere prioritaria rispetto alla partecipazione all’esercizio fisico [16]. Tuttavia, l’ingestione orale di BCAA è molto discutibile per quanto riguarda l’ottimizzazione delle prestazioni e della sintesi proteica [14,17,18]. Recenti review sistematiche mostrano che l’integrazione di BCAA tende ad attenuare l’indolenzimento muscolare, che è un indicatore di danno muscolare [17,18]. Gli effetti ergogenici dei BCAA sono stati esaminati principalmente negli adulti e pochi studi hanno incluso partecipanti allenati, che possono differire sostanzialmente in termini di prestazioni, danno muscolare e composizione corporea. Inoltre, l’Australian Institute of Sport ha classificato i BCAA nel gruppo C, che comprende gli integratori privi di supporto scientifico tra gli atleti o studi non conclusivi. Tuttavia, i BCAA non sono raccomandati nei programmi di integrazione [15]. La panoramica del Comitato Olimpico Internazionale non ha menzionato i BCAA tra i diversi argomenti trattati, ovvero gli integratori utilizzati per prevenire o trattare le carenze di nutrienti, gli integratori utilizzati per fornire energia e gli integratori che migliorano le prestazioni sportive [19]. Per quanto riguarda i tre BCAA, sono stati descritti solo gli effetti della leucina sul turnover proteico, anche se gli studi citati non includevano partecipanti allenati [20,21].
Negli studi che hanno valutato l’impatto dell’integrazione di BCAA sulle prestazioni, i partecipanti erano in genere atleti di ciclismo e corsa [21,22,24,26], atleti impegnati in allenamenti contro-resistenza [23,25,27], pallavolisti [28] e calciatori [29,30]. Tenendo conto delle variazioni nei protocolli di integrazione, i risultati suggeriscono che i BCAA hanno effetti insignificanti sulle prestazioni. In un altro studio, i partecipanti hanno seguito una dieta ipocalorica con carboidrati o BCAA e un pesante programma di allenamento contro-resistenza per 8 settimane [26]. Gli autori hanno riscontrato che l’integrazione con BCAA ha aumentato significativamente la forza della parte superiore (15,1 ± 2,2 kg) e inferiore del corpo (7,1 ± 1,6 kg), mentre il gruppo CHO ha mostrato cambiamenti trascurabili nella forza della parte superiore del corpo (4,8 ± 1,8 kg). Uno studio separato, che ha esaminato gli effetti dell’integrazione di BCAA a lungo termine per 10 settimane in 18 ciclisti, ha riscontrato un’interazione tra gruppo e tempo per il picco di potenza nel gruppo BCAA [27]. Tuttavia, gli autori non hanno controllato l’apporto nutrizionale e gli atleti sono stati istruiti a mantenere le loro abitudini alimentari durante l’indagine [27]. Altri studi hanno esaminato gli effetti dell’integrazione di BCAA per 7-8 giorni [25,31,32] immediatamente prima, durante o dopo l’esercizio fisico [21,28,29,30]. Per questi studi, pochi manoscritti hanno testato l’integrazione di BCAA in riferimento agli indicatori di prestazione.
L’efficacia dell’integrazione di BCAA sulla composizione corporea è stata esaminata in 50 corridori amatoriali [25], 17 atleti contro-resistenza [26] e 18 ciclisti [27]. Dopo 7 giorni di integrazione orale, sono stati osservati cambiamenti comparabili nel peso corporeo nel gruppo BCAA e nel gruppo di controllo [25]. Sono stati osservati effetti trascurabili per il gruppo BCAA nel tessuto magro e nella massa grassa [26,27].
L’indolenzimento o il recupero muscolare sono stati valutati mediante l’uso di scale o esaminando i cali di prestazione post-esercizio attraverso l’uso di test specifici sul campo o in laboratorio. Negli sport di resistenza (corsa o ciclismo), i risultati degli studi estratti non sono conclusivi. Le valutazioni dello sforzo percepito e della fatica mentale sono risultate significativamente ridotte nel gruppo BCAA tra sette ciclisti di resistenza [21], mentre in 50 maratoneti non sono state riportate differenze nelle valutazioni dello sforzo percepito [25]. Al contrario, l’indolenzimento muscolare è risultato inferiore nel gruppo BCAA rispetto al placebo in 16 corridori di distanza [33]. Gli studi che hanno incluso partecipanti allenati contro-resistenza e che hanno esaminato gli effetti dei BCAA sull’indolenzimento muscolare o sul recupero hanno indicato potenziali benefici di leucina, isoleucina o valina nell’attenuazione dell’indolenzimento muscolare dopo l’esercizio [34,35]. Due studi hanno riportato che il consumo di un’integrazione di BCAA ha attenuato i cali di prestazione [36,37]. Una serie di parametri biochimici, ormonali e molecolari sono stati analizzati dopo un intervento di esercizio fisico combinato con l’integrazione di BCAA. I risultati principali di questi studi estratti rivelano che l’assunzione di BCAA ha causato un sostanziale miglioramento del rapporto BCAA:triptofano [21,27] e della risposta immunitaria [27,38,39]. Tenendo conto delle variazioni metodologiche tra gli studi, i cambiamenti ormonali indicano che l’integrazione di BCAA ha favorito una risposta ormonale anabolizzante. Il livello di cortisolo è diminuito dopo l’esercizio fisico e, parallelamente, il testosterone tendeva ad aumentare nei partecipanti impegnati nell’allenamento contro-resistenza [40]. Gli studi che hanno esaminato i meccanismi biochimici si sono concentrati principalmente su segnali metabolici specifici e sulla loro dipendenza dal meccanismo del complesso target della rapamicina 1 (mTORC1).
BCAA e sintesi proteica muscolare:
Le proteine muscolari sono in costante stato di turnover, il che significa che vengono continuamente prodotte nuove proteine mentre quelle più vecchie vengono degradate. Lo stato anabolico non ha una definizione specifica, ma in generale si riferisce alla circostanza in cui il tasso di sintesi delle proteine muscolari supera il tasso di degradazione delle proteine muscolari. Il risultato è un aumento della massa muscolare. Convenzionalmente si ritiene che lo stato anabolico sia guidato da una stimolazione della sintesi proteica muscolare, ma teoricamente potrebbe anche derivare da un’inibizione della degradazione delle proteine muscolari.
L’obiettivo metabolico principale del consumo di integratori di BCAA è quello di massimizzare lo stato anabolico. È opinione diffusa che i BCAA inducano uno stato anabolico stimolando la sintesi proteica muscolare. Un’abbondante disponibilità di tutti gli EAA è un requisito per una stimolazione significativa della sintesi proteica muscolare [41]. La sintesi proteica muscolare sarà limitata dalla mancanza di disponibilità di uno qualsiasi degli EAA, mentre una carenza di NEAA può essere compensata da una maggiore produzione de novo dei NEAA carenti [41]. Nello stato postprandiale successivo a un pasto contenente proteine, tutti i precursori degli EAA necessari per la sintesi di nuove proteine muscolari possono essere ricavati dalle elevate concentrazioni plasmatiche derivanti dalla digestione delle proteine consumate o dal riciclo dalla scomposizione delle proteine. In questa circostanza di abbondante disponibilità di EAA, il tasso di sintesi proteica muscolare supera il tasso di degradazione, producendo così uno stato anabolico. Nello stato post-assorbitivo i livelli plasmatici di EAA scendono al di sotto dei valori post-prandiali perché gli aminoacidi non vengono più assorbiti. Di conseguenza, gli EAA non vengono più assunti dal muscolo, ma rilasciati dal muscolo nel plasma [42]. Questo stato catabolico della proteina muscolare nello stato post-assorbitivo consente di mantenere la disponibilità di EAA per altri tessuti per mantenere il tasso di sintesi proteica a spese della proteina muscolare, che può essere considerata come una riserva di EAA a cui attingere per il resto del corpo.
Poiché gli EAA non possono essere prodotti nell’organismo e vi è un rilascio netto di EAA dal muscolo, nello stato post-assorbitivo l’unica fonte di precursori di EAA per la sintesi proteica muscolare è costituita dagli EAA intracellulari derivati dalla degradazione delle proteine muscolari [42]. Oltre a essere reincorporati nelle proteine muscolari attraverso la sintesi, alcuni EAA rilasciati dalla disgregazione delle proteine muscolari possono essere parzialmente ossidati all’interno del muscolo, rendendoli così indisponibili per la reincorporazione nelle proteine muscolari. Gli EAA rilasciati dalla degradazione delle proteine muscolari che non vengono reincorporati nelle proteine muscolari o ossidati all’interno del tessuto muscolare vengono rilasciati nel plasma, dove possono essere assorbiti da altri tessuti come precursori per la sintesi proteica o ossidati irreversibilmente [43]. Pertanto, il tasso di sintesi proteica muscolare sarà sempre inferiore al tasso di degradazione delle proteine muscolari nello stato post-assorbitivo, a causa del flusso netto di EAA dalla degradazione delle proteine al plasma e alle vie ossidative. In altre parole, è impossibile che la sintesi proteica muscolare superi il tasso di degradazione delle proteine muscolari quando i precursori derivano interamente dalla degradazione delle proteine, e quindi non si può verificare uno stato anabolico in assenza di assunzione di aminoacidi esogeni.
Tutti i precursori EAA per la sintesi proteica muscolare nello stato post-assorbitivo derivano dalla disgregazione delle proteine muscolari. È stato costantemente riportato che negli esseri umani normali in fase post-assorbitiva il tasso di degradazione delle proteine muscolari supera il tasso di sintesi delle proteine muscolari di circa il 30% [44]. Il consumo dei soli BCAA (cioè senza gli altri EAA) può aumentare la sintesi proteica muscolare nello stato post-assorbitivo solo aumentando l’efficienza del riciclo degli EAA dalla disgregazione proteica alla sintesi proteica, invece di essere rilasciati nel plasma o ossidati. Questo perché tutti i 9 EAA (e gli 11 NEAA) sono necessari per produrre proteine muscolari e gli EAA non possono essere prodotti dall’organismo. Se si consumano solo 3 EAA, come nel caso del consumo di BCAA, la ripartizione proteica è l’unica fonte dei restanti EAA necessari come precursori per la sintesi proteica muscolare. È quindi teoricamente impossibile che il consumo di soli BCAA crei uno stato anabolico in cui la sintesi proteica muscolare superi la degradazione delle proteine muscolari. Se si ipotizza generosamente che il consumo di BCAA migliori del 50% l’efficienza del riciclo degli EAA dalla disgregazione delle proteine muscolari alla sintesi delle proteine muscolari, ciò si tradurrebbe in un aumento del 15% del tasso di sintesi delle proteine muscolari (30% riciclato allo stato basale X 50% miglioramento del riciclo = 15% aumento della sintesi). Inoltre, una riduzione del 50% del rilascio di EAA nel plasma dal muscolo ridurrebbe anche i pool plasmatici e intracellulari di EAA liberi. La figura seguente illustra schematicamente questi principi. Poiché un miglioramento del 50% nell’efficienza del riciclo sarebbe circa il limite massimo ragionevole, ciò significa che la stimolazione massima della sintesi proteica muscolare non potrebbe superare il 15%. Ciò corrisponderebbe a un aumento del tasso di sintesi frazionale del muscolo da un valore di circa 0,050%/h allo stato basale a 0,057%/h, e questa differenza nel tasso di sintesi frazionale (FSR) delle proteine sarebbe difficile da misurare con precisione [45].
Rappresentazione schematica del riciclo degli aminoacidi essenziali (EAA) dalla disgregazione delle proteine muscolari alla sintesi delle proteine muscolari nello stato post-assorbitivo. Le unità arbitrarie sono utilizzate per semplicità e si basano sui tassi misurati di ciascuna via in soggetti umani in fase post-assorbitiva in circostanza normale nello stato post-assorbitivo. Circa il 70% degli EAA provenienti dalla disgregazione delle proteine muscolari viene riciclato nella sintesi proteica. Esiste un efflusso netto di circa l’85% degli EAA rilasciati dalla disgregazione proteica, che possono essere assunti e incorporati nelle proteine di altri tessuti oppure ossidarsi. Circa il 15% degli EAA provenienti dalla degradazione delle proteine viene parzialmente ossidato nel muscolo e non è disponibile per la sintesi proteica. Le cifre relative al flusso verso l’esterno e all’ossidazione intracellulare degli EAA sono medie, poiché alcuni EAA, come la fenilalanina, non vengono ossidati affatto nel muscolo. b Rappresentazione di un aumento del 50% dell’efficienza del riciclo degli EAA dalla disgregazione delle proteine muscolari alla sintesi proteica. In questo esempio si avrebbe un aumento della sintesi da 70 a 80 unità, ovvero del 20%. La sintesi proteica non può mai superare la ripartizione proteica nello stato post-assorbitivo, poiché la ripartizione proteica è l’unica fonte di EAA.
I BCAA sono stati somministrati per via endovenosa negli unici studi che hanno determinato la risposta del metabolismo proteico muscolare in soggetti umani ai soli BCAA. Sebbene l’infusione di BCAA non sia la modalità convenzionale di assunzione di un integratore alimentare, è stato dimostrato che gli aminoacidi infusi per via endovenosa e quelli ingeriti per via orale producono effetti comparabili sulla sintesi proteica muscolare in altre circostanze [46]. Di conseguenza, è ragionevole valutare i documenti in cui viene descritta la risposta della sintesi proteica muscolare all’infusione endovenosa di BCAA in soggetti umani.
Louard et al. [47] hanno utilizzato il metodo dell’equilibrio dell’avambraccio per quantificare la risposta all’infusione endovenosa di una miscela di BCAA per 3 ore in 10 soggetti in fase post-assorbitiva. Il metodo dell’equilibrio dell’avambraccio prevede la misurazione dell’assorbimento e del rilascio di singoli EAA (leucina e fenilalanina in questo caso) e delle loro controparti marcate isotopicamente. Vengono calcolati i tassi di scomparsa (Rd) e di comparsa (Ra) di fenilalanina e leucina. Partendo dal presupposto che il bilancio di leucina e fenilalanina nel muscolo è rappresentativo di tutti gli EAA, il Rd. della fenilalanina è considerato un riflesso della sintesi proteica muscolare, poiché la sintesi proteica è l’unico destino della fenilalanina assunta dal plasma nel muscolo. La Rd. della leucina non può essere interpretata in relazione alla sintesi proteica, poiché la leucina assunta dal muscolo può essere ossidata oltre che incorporata nelle proteine. L’infusione di 3 ore di BCAA ha aumentato le concentrazioni plasmatiche di tutti e 3 i BCAA di quattro volte, mentre le concentrazioni di altri EAA sono diminuite [47]. Invece di essere stimolata dall’infusione di BCAA, la sintesi proteica muscolare è diminuita da 37+/- 3 a 21 +/- 2 nmol/min/100 ml di gamba (statisticamente significativo, p < 0,05) [47]. Non si sono verificate variazioni significative nel bilancio netto della fenilalanina, il che indica che anche la degradazione delle proteine muscolari è stata ridotta in misura simile alla riduzione della sintesi proteica muscolare. Il bilancio tra la sintesi e la degradazione delle proteine muscolari è rimasto negativo, il che significa che lo stato catabolico è persistito e non si è prodotto uno stato anabolico. La diminuzione simultanea della sintesi e della degradazione delle proteine muscolari durante l’infusione di BCAA può essere descritta come una diminuzione del turnover proteico muscolare.
Risultati simili sono stati ottenuti dagli stessi ricercatori quando hanno esteso l’infusione di BCAA a 16 ore in 8 volontari normali e hanno determinato se l’aumento cronico di BCAA stimolasse la sintesi proteica muscolare [48]. Per calcolare la sintesi e la ripartizione delle proteine muscolari è stata utilizzata la stessa metodologia di bilanciamento dell’avambraccio dello studio precedente. L’infusione di 16 ore ha aumentato le concentrazioni di BCAA da 5 a 8 volte [48], ovvero il doppio dei livelli raggiunti con una dose normale di BCAA ingeriti per via orale [49]. Come nello studio precedente, la sintesi proteica muscolare (riflessa dalla fenilalanina Rd) si è ridotta nei soggetti che hanno ricevuto i BCAA rispetto all’infusione di soluzione salina, passando da 36 +/- 5 a 27 +/-2 nmol/min/100 ml. Anche la disgregazione proteica muscolare si è ridotta, il che significa che anche il turnover proteico muscolare è stato ridotto e che è persistito uno stato catabolico.
Da questi due studi possiamo concludere che l’infusione di BCAA non solo non aumenta il tasso di sintesi proteica muscolare nei soggetti umani, ma anzi riduce il tasso di sintesi proteica muscolare e il tasso di turnover proteico muscolare. Lo stato catabolico non è stato invertito in uno stato anabolico in nessuno dei due studi. Inoltre, una riduzione prolungata del tasso di turnover delle proteine muscolari dovrebbe avere un effetto negativo sulla forza muscolare, anche se la massa muscolare viene mantenuta. Il ricambio delle proteine muscolari rinnova le fibre muscolari e determina una maggiore efficienza della contrazione a livello di singola fibra [50], che si riflette in un aumento della forza in vivo, indipendentemente dalla massa muscolare [51, 52].
Il mancato aumento significativo della sintesi proteica muscolare in risposta all’infusione dei soli BCAA è atteso in base alle considerazioni teoriche discusse in precedenza e illustrate nella figura sopra esposta per quanto riguarda il requisito di tutti gli EAA per sostenere un aumento. Invece, poiché la disgregazione delle proteine muscolari è diminuita, è diminuita anche la disponibilità di EAA, che a sua volta ha ridotto il tasso di sintesi proteica muscolare.
L’affermazione che la sintesi proteica muscolare sia stimolata dai BCAA deriva, almeno in parte, dall’osservazione dell’aumento della segnalazione anabolica intracellulare, compreso lo stato di attivazione di fattori chiave coinvolti nell’avvio della sintesi proteica [53]. La teoria secondo cui l’attivazione dei fattori di segnalazione anabolica intracellulare provoca un aumento del tasso di sintesi proteica muscolare si è radicata nei moderni concetti di regolazione della sintesi proteica muscolare. L’aumento della segnalazione anabolica in risposta ai BCAA è stato citato come prova di una stimolazione della sintesi proteica muscolare, anche in assenza della misurazione della sintesi proteica muscolare (ad esempio, [53]). Tuttavia, l’attivazione delle vie di segnalazione anabolica può coincidere con un aumento della sintesi proteica muscolare solo in presenza di un’ampia quantità di EAA che forniscano i precursori necessari per produrre proteine complete.
La dissociazione tra lo stato di fosforilazione dei fattori di segnalazione e la sintesi proteica muscolare nell’uomo è stata dimostrata in diverse circostanze quando la disponibilità di tutti gli EAA è limitata. Ad esempio, un aumento della concentrazione di insulina (ad esempio in seguito all’assunzione di glucosio) è un potente attivatore delle vie di segnalazione anabolica, ma non riesce ad aumentare la FSR muscolare a causa della carenza di EAA [54]. Al contrario, il consumo di una piccola quantità (3 g) di EAA stimola la sintesi proteica muscolare senza influenzare l’attività dei fattori di iniziazione, come Akt, S6 chinasi e 4E-BP1 [55]. Un piccolo aumento delle concentrazioni plasmatiche di EAA non avrebbe alcun effetto se la sintesi proteica fosse limitata dallo stato di attivazione dei fattori di iniziazione. Negli studi citati in precedenza, in cui i BCAA sono stati infusi per via endovenosa, è ragionevole presumere che un aumento così consistente delle concentrazioni di BCAA avrebbe attivato i fattori di segnalazione, eppure la sintesi proteica muscolare è effettivamente diminuita a causa della mancanza di disponibilità di EAA derivante da una diminuzione della disgregazione proteica. Pertanto, nei soggetti umani la somministrazione di EAA può aumentare la sintesi proteica muscolare in assenza di qualsiasi cambiamento nell’attivazione dei fattori di iniziazione, mentre l’attivazione dei fattori di iniziazione in assenza del consumo di tutti gli EAA non ha alcun effetto sulla sintesi proteica muscolare. Questi risultati possono essere interpretati solo come la dimostrazione che il controllo limitante della sintesi proteica muscolare basale nell’uomo è la disponibilità di tutti gli EAA e non l’attività dei fattori di segnalazione anabolica. Questa conclusione mette ulteriormente in dubbio il ruolo dell’integrazione alimentare dei soli BCAA come stimolatori della sintesi proteica muscolare.
Se si considerano tutte le prove e le teorie, è ragionevole concludere che non esistono prove credibili che l’ingestione di un integratore alimentare di BCAA determini da solo una stimolazione fisiologicamente significativa delle proteine muscolari. Anzi, le prove disponibili indicano che i BCAA in realtà diminuiscono la sintesi proteica muscolare. Tutti gli EAA devono essere disponibili in abbondanza perché l’aumento della segnalazione anabolica si traduca in un’accelerazione della sintesi proteica muscolare.
A differenza della mancanza di un effetto interattivo tra BCAA e carboidrati, i BCAA possono potenziare l’effetto anabolico di un pasto proteico. Ad esempio, l’aggiunta di 5 g di BCAA a una bevanda contenente 6,25 g di proteine del siero di latte ha aumentato la sintesi proteica muscolare a un livello paragonabile a quello indotto da 25 g di proteine del siero di latte [56]. Questo risultato suggerisce che uno o più BCAA potrebbero essere limitanti per la stimolazione della sintesi proteica muscolare da parte delle proteine del siero di latte, oppure che i BCAA in più inducono un maggiore potenziale di risposta anabolica del muscolo alle proteine del siero di latte attivando i fattori di iniziazione. In entrambi i casi, la risposta dei BCAA in combinazione con le proteine intatte è una questione diversa rispetto all’effetto dei soli BCAA, poiché le proteine intatte forniscono tutti gli EAA necessari per produrre una proteina intatta.
Le risposte ai singoli BCAA (cioè leucina, valina o isoleucina) potrebbero differire dalla combinazione dei tre per diversi motivi. È dimostrato che la leucina da sola può esercitare una risposta anabolica (ad esempio, [57]), mentre non esistono dati simili per l’isoleucina o la valina. Pertanto, ci si potrebbe aspettare che la leucina da sola sia più efficace della combinazione di tutti i BCAA. Tuttavia, un’integrazione alimentare di sola leucina presenta due limitazioni significative. In primo luogo, gli stessi problemi che limitano l’entità della stimolazione della sintesi proteica muscolare da parte dei soli BCAA, relativi alla disponibilità degli altri EAA necessari per la produzione di proteine muscolari intatte, limitano anche la risposta alla sola leucina. In secondo luogo, l’aumento della concentrazione plasmatica di leucina attiva la via metabolica che ossida tutti i BCAA. Di conseguenza, l’ingestione della sola leucina determina una diminuzione delle concentrazioni plasmatiche di valina e isoleucina. La disponibilità di valina e isoleucina può quindi diventare limitante per la sintesi proteica muscolare quando si consuma solo leucina. Questo potrebbe essere il motivo per cui gli studi sui risultati a lungo termine con l’integrazione dietetica di leucina non hanno dato risultati positivi [58]. Il motivo principale per cui un integratore alimentare contiene tutti i BCAA rispetto alla sola leucina è quello di superare le diminuzioni delle concentrazioni plasmatiche di valina e isoleucina che si verificherebbero quando la leucina viene somministrata da sola.
Sebbene un integratore alimentare con tutti i BCAA superi le diminuzioni di concentrazione derivanti dal consumo della sola leucina, l’aggiunta di valina e isoleucina può comunque limitare l’efficacia della sola leucina a causa della competizione per il trasporto nelle cellule muscolari. I BCAA sono tutti trasportati attivamente nelle cellule, comprese quelle muscolari, dallo stesso sistema di trasporto. Pertanto, se forniti insieme, i BCAA competono tra loro per il trasporto nelle cellule. Se uno dei BCAA (ad esempio, la leucina) è limitante per la sintesi proteica, l’aggiunta degli altri due BCAA potrebbe limitare la stimolazione della sintesi proteica a causa del ridotto ingresso della leucina nella cellula. I BCAA competono anche con altri aminoacidi per il trasporto, compresa la fenilalanina, e questa competizione potrebbe influenzare la disponibilità intramuscolare di altri EAA. A causa della competizione per i trasportatori, è possibile che la leucina da sola, ad esempio, abbia un effetto stimolante transitorio sulla sintesi proteica muscolare (ad esempio, [59]) laddove i BCAA non riescono a suscitare tale risposta [60, 61].
Conclusioni:
Come abbiamo visto, la ricerca ha evidenziato che l’integrazione di BCAA non sembra avere un impatto significativo sulle prestazioni. D’altra parte, l’ingestione orale di BCAA isolati riduce l’indolenzimento muscolare. I BCAA sono disponibili anche in diversi prodotti di integrazione (ad esempio, proteine del siero del latte) e sono spesso combinati con altri nutrienti (ad esempio, carboidrati). Pertanto, i potenziali benefici dell’integrazione di BCAA isolati negli atleti per attenuare l’indolenzimento muscolare e ritardare l’affaticamento devono essere interpretati con cautela.
Rimane il fatto che un aumento fisiologicamente significativo del tasso di sintesi proteica muscolare richiede un’adeguata disponibilità di tutti i precursori aminoacidici. La fonte di EAA per la sintesi proteica muscolare nello stato post-assorbitivo è il pool libero intracellulare. Gli EAA liberi intracellulari disponibili per l’incorporazione nelle proteine derivano dalla degradazione delle proteine muscolari. In condizioni normali, circa il 70% degli EAA rilasciati dalla degradazione delle proteine muscolari viene reincorporato nelle proteine muscolari. L’efficienza della reincorporazione degli EAA provenienti dalla degradazione delle proteine nelle proteine muscolari può essere aumentata solo in misura limitata. Per questo motivo fondamentale, un’integrazione alimentare di soli BCAA non può sostenere un aumento del tasso di sintesi proteica muscolare. La disponibilità degli altri EAA diventerà rapidamente limitante per la sintesi proteica accelerata. Coerentemente con questa prospettiva, i pochi studi condotti su soggetti umani hanno riportato una diminuzione, piuttosto che un aumento, della sintesi proteica muscolare dopo l’assunzione di BCAA. Si può concludere, quindi, che gli integratori alimentari di BCAA da soli non promuovono l’anabolismo muscolare.
Direi che le informazioni riportate siano sufficienti a far desistere nell’acquisto di questo integratore chiunque sia dotato di un minimo di capacità cognitiva…
Jump up to:ab Scaini, G.; Jeremias, I. C.; Morais, M. O.; Borges, G. D.; Munhoz, B. P.; Leffa, D. D.; Andrade, V. M.; Schuck, P. F.; Ferreira, G. C.; Streck, E. L. (2012). “DNA damage in an animal model of maple syrup urine disease”. Molecular Genetics and Metabolism. 106 (2): 169–174. doi:10.1016/j.ymgme.2012.04.009. PMID22560665.
Jump up to:ab Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, et al. (November 2001). “Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo”. Nature Cell Biology. 3 (11): 1014–9. doi:10.1038/ncb1101-1014. PMID11715023. S2CID16284975.
Phillips S.M. The impact of protein quality on the promotion of resistance exercise-induced changes in muscle mass. Nutr. Metab. 2016;13:64. doi: 10.1186/s12986-016-0124-8. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Plotkin D.L., Delcastillo K., Van Every D.W., Tipton K.D., Aragon A.A., Schoenfeld B.J. Isolated Leucine and Branched-Chain Amino Acid Supplementation for Enhancing Muscular Strength and Hypertrophy: A Narrative Review. Int. J. Sport Nutr. Exerc. Metab. 2021;31:292–301. doi: 10.1123/ijsnem.2020-0356. [PubMed] [CrossRef] [Google Scholar]
Australian Institute of Sports . Branched-Chain Amino Acids (Bcaa) Summary Report: Consideration for Classification of a Supplement Ingredient. Australian Institute of Sport; Bruce, Australia: 2021. [Google Scholar]
Jäger R., Kerksick C.M., Campbell B.I., Cribb P.J., Wells S.D., Skwiat T.M., Purpura M., Ziegenfuss T.N., Ferrando A.A., Arent S.M., et al. International Society of Sports Nutrition Position Stand: Protein and exercise. J. Int. Soc. Sports Nutr. 2017;14:20. doi: 10.1186/s12970-017-0177-8. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Wolfe R.R. Branched-chain amino acids and muscle protein synthesis in humans: Myth or reality? J. Int. Soc. Sports Nutr. 2017;14:30. doi: 10.1186/s12970-017-0184-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Doma K., Singh U., Boullosa D., Connor J.D. The effect of branched-chain amino acid on muscle damage markers and performance following strenuous exercise: A systematic review and meta-analysis. Appl. Physiol. Nutr. Metab. 2021;46:1303–1313. doi: 10.1139/apnm-2021-0110. [PubMed] [CrossRef] [Google Scholar]
Hormoznejad R., Zare A.J., Mansoori A. Effect of BCAA supplementation on central fatigue, energy metabolism substrate and muscle damage to the exercise: A systematic review with meta-analysis. Sport Sci. Health. 2019;15:265–279. doi: 10.1007/s11332-019-00542-4. [CrossRef] [Google Scholar]
Maughan R.J., Burke L.M., Dvorak J., Larson-Meyer D.E., Peeling P., Phillips S.M., Rawson E.S., Walsh N.P., Garthe I., Geyer H., et al. IOC consensus statement: Dietary supplements and the high-performance athlete. Br. J. Sports Med. 2018;52:439–455. doi: 10.1136/bjsports-2018-099027. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Wilkinson D.J., Hossain T., Hill D.S., Phillips B.E., Crossland H., Williams J., Loughna P., Churchward-Venne T.A., Breen L., Phillips S.M., et al. Effects of leucine and its metabolite β-hydroxy-β-methylbutyrate on human skeletal muscle protein metabolism. J. Physiol. 2013;591:2911–2923. doi: 10.1113/jphysiol.2013.253203. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Areces F., Salinero J.J., Abian-Vicen J., González-Millán C., Gallo-Salazar C., Ruiz-Vicente D., Lara B., Del Coso J. A 7-day oral supplementation with branched-chain amino acids was ineffective to prevent muscle damage during a marathon. Amino Acids. 2014;46:1169–1176. doi: 10.1007/s00726-014-1677-3. [PubMed] [CrossRef] [Google Scholar]
Dudgeon W.D., Kelley E.P., Scheett T.P. In a single-blind, matched group design: Branched-chain amino acid supplementation and resistance training maintains lean body mass during a caloric restricted diet. J. Int. Soc. Sports Nutr. 2016;13:1. doi: 10.1186/s12970-015-0112-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Kephart W.C., Wachs T.D., Thompson R.M., Brooks Mobley C., Fox C.D., McDonald J.R., Ferguson B.S., Young K.C., Nie B., Martin J.S., et al. Ten weeks of branched-chain amino acid supplementation improves select performance and immunological variables in trained cyclists. Amino Acids. 2016;48:779–789. doi: 10.1007/s00726-015-2125-8. [PubMed] [CrossRef] [Google Scholar]
Moberg M., Apró W., Ekblom B., van Hall G., Holmberg H.C., Blomstrand E. Activation of mTORC1 by leucine is potentiated by branched-chain amino acids and even more so by essential amino acids following resistance exercise. Am. J. Physiol. Cell Physiol. 2016;310:C874–C884. doi: 10.1152/ajpcell.00374.2015. [PubMed] [CrossRef] [Google Scholar]
Samuelsson H., Moberg M., Apró W., Ekblom B., Blomstrand E. Intake of branched-chain or essential amino acids attenuates the elevation in muscle levels of PGC-1α4 mRNA caused by resistance exercise. Am. J. Physiol. Endocrinol. Metab. 2016;311:E246–E251. doi: 10.1152/ajpendo.00154.2016. [PubMed] [CrossRef] [Google Scholar]
Smith J.W., Krings B.M., Shepherd B.D., Waldman H.S., Basham S.A., McAllister M.J. Effects of carbohydrate and branched-chain amino acid beverage ingestion during acute upper body resistance exercise on performance and postexercise hormone response. Appl. Physiol. Nutr. Metab. 2018;43:504–509. doi: 10.1139/apnm-2017-0563. [PubMed] [CrossRef] [Google Scholar]
Martín-Martínez J.P., Calleja Gonzalez J., Adsuar Sala J.C., Gómez-Pomares S., Carlos-Vivas J., Pérez-Gómez J. Short-term branched-chain amino acid supplementation does not enhance vertical jump in professional volleyball players. A double-blind, controlled, randomized study. Nutr. Hosp. 2020;37:1007–1011. [PubMed] [Google Scholar]
Pancar S. The effect of Branched Chain Amino Acids Intake Before and after Exercise on Physical Performance and Recovery. Ambient. Sci. 2020;7:243–246. doi: 10.21276/ambi.2020.07.sp1.oa
[CrossRef] [Google Scholar]33. Mor A., Acar K., Yilmaz A.K., Arslanoglu E. The effects of BCAA and creatine supplementation on anaerobic capacity and ball kicking speed in male football players. J. Mens Health. 2022;18:5. [Google Scholar]34. Koba T., Hamada K., Samurai M., Matsumoto K., Higuchi T., Zhao M., Miyata H. Effect of A Branched-chain Amino Acids Supplementation on Muscle Soreness during Intensive Training Program: 229: Board# 136: 11:00 AM–12:30 PM. Med. Sci. Sports Exerc. 2005;37:S43. [Google Scholar]
Gee T.I., Deniel S. Branched-chain aminoacid supplementation attenuates a decrease in power-producing ability following acute strength training. J. Sports Med. Phys. Fit. 2016;56:1511–1517. [PubMed] [Google Scholar]
Waldron M., Whelan K., Jeffries O., Burt D., Howe L., Patterson S.D. The effects of acute branched-chain amino acid supplementation on recovery from a single bout of hypertrophy exercise in resistance-trained athletes. Appl. Physiol. Nutr. Metab. 2017;42:630–636. doi: 10.1139/apnm-2016-0569. [PubMed] [CrossRef] [Google Scholar]
VanDusseldorp T.A., Escobar K.A., Johnson K.E., Stratton M.T., Moriarty T., Cole N., McCormick J.J., Kerksick C.M., Vaughan R.A., Dokladny K., et al. Effect of Branched-Chain Amino Acid Supplementation on Recovery Following Acute Eccentric Exercise. Nutrients. 2018;10:1389. doi: 10.3390/nu10101389. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Bassit R.A., Sawada L.A., Bacurau R.F., Navarro F., Martins E., Jr., Santos R.V., Caperuto E.C., Rogeri P., Costa Rosa L.F. Branched-chain amino acid supplementation and the immune response of long-distance athletes. Nutrition. 2002;18:376–379. doi: 10.1016/S0899-9007(02)00753-0. [PubMed] [CrossRef] [Google Scholar]
Bassit R.A., Sawada L.A., Bacurau R.F., Navarro F., Costa Rosa L.F. The effect of BCAA supplementation upon the immune response of triathletes. Med. Sci. Sports Exerc. 2000;32:1214–1219. doi: 10.1097/00005768-200007000-00005. [PubMed] [CrossRef] [Google Scholar]
Nemet D., Wolach B., Eliakim A. Proteins and amino acid supplementation in sports: Are they truly necessary? Isr. Med. Assoc. J. 2005;7:328–332. [PubMed] [Google Scholar]
Vahid I., Abdolali B., Fatemeh M., Alireza N., Mehdi S. The effects of branch-chain amino acids on fatigue in the athletes. Interv. Med. Appl. Sci. 2018;10:233–235. doi: 10.1556/1646.10.2018.10. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Lysenko E.A., Vepkhvadze T.F., Lednev E.M., Vinogradova O.L., Popov D.V. Branched-chain amino acids administration suppresses endurance exercise-related activation of ubiquitin proteasome signaling in trained human skeletal muscle. J. Physiol. Sci. 2018;68:43–53. doi: 10.1007/s12576-016-0506-8. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Goldberg A.L., Chang T.W. Regulation and significance of amino acid metabolism in skeletal muscle. Fed. Proc. 1978;37:2301–2307. [PubMed] [Google Scholar]
Stipanuk M.H., Claudill M.A. Biochemical, Physiological, and Molecular Aspects of Human Nutrition. Saunders; St. Louis, MO, USA: 2013. [Google Scholar]
Volpi E, Kobayashi H, Sheffield-Moore M, Mittendorfer B, Wolfe RR. Essential amino acids are primarily responsible for the amino acid stimulation of muscle protein anabolism in healthy elderly adults. Am J Clin Nutr. 2003;78:250–258. [PMC free article] [PubMed] [Google Scholar]
Wolfe RR. The underappreciated role of muscle in health and disease. Am J Clin Nutr. 2006;84(3):475–482. [PubMed] [Google Scholar]
Biolo G, Gastaldelli A, Zhang X-J, Wolfe RR. Protein synthesis and breakdown in skin and muscle: a leg model of amino acid kinetics. Am J Physiol Endocrinol Metab. 1994;30:E467–E474. [PubMed] [Google Scholar]
Smith GI, Patterson BW, Mittendorfer B. Human muscle protein turnover-why is it so variable? J Appl Physiol. 1985;110:480–491. doi: 10.1152/japplphysiol.00125.2010. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Rasmussen BB, Wolfe RR, Volpi E. Oral and intravenously administered amino acids produce similar effects on muscle protein synthesis in the elderly. J Nutr Health Aging. 2002;6:358–362. [PMC free article] [PubMed] [Google Scholar]
Louard RJ, Barrett EJ, Gelfand RA. Effect of infused branched-chain amino acids on muscle and whole body amino acid metabolism in man. Clin Sci. 1990;79:457–466. doi: 10.1042/cs0790457. [PubMed] [CrossRef] [Google Scholar]
Matthews DE. Observations of branched-chain amino acid administration in humans. J Nutr. 2005;135:1580S–1584S. [PMC free article] [PubMed] [Google Scholar]
Fitts RH, Ramatowski JG, Peters JR, Paddon-Jones D, Wolfe RR, Ferrando AA. The deleterious effects of bed rest on human skeletal muscle fibers are exacerbated by hypercortisolemia and ameliorated by dietary supplementation. Am J Physiol Cell Physiol. 2007;293:C313–C320. doi: 10.1152/ajpcell.00573.2006. [PubMed] [CrossRef] [Google Scholar]
Balagopal P, Rooyackers OE, Adey DB, Ades PA, Nair KS. Effects of aging on in vivo synthesis of skeletal muscle myosin heavy-chain and sarcoplasmic protein in humans. Am J Phys. 1997;273:E790–E800. [PubMed] [Google Scholar]
Paddon-Jones D, Sheffield-Moore M, Urban RJ, Aarsland A, Wolfe RR, Ferrando AA. The catabolic effects of prolonged inactivity and acute hypercortisolemia are offset by dietary supplementation. J Clin Endocrinol Metab. 2005;90(3):1453–1459. doi: 10.1210/jc.2004-1702. [PubMed] [CrossRef] [Google Scholar]
Blomstrand E, Eliasson J, Karlsson HKR, Kohnke R. Branched-chain amino acids activate key enzymes in protein synthesis after physical exercise. J Nutr. 2006;136:269S–273S. [PubMed] [Google Scholar]
Greenhaff PL, Karagounis LG, Peirce N, Simpson EJ, Hazell M, Layfield R, Wackerhage H, Smith K, Atherton P, Selby A, Rennie MJ. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in muscle. Am J Physiol Endocrinol Metab. 2008;295:E 590–E 597. doi: 10.1152/ajpendo.90411.2008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Bukari SS, Phillips BE, Wilinson DJ, Limb MC, Rankin D, Mitchell WK, Kobayashi H, Greenhaff PL, Smith K, Atherton PJ. Intake of low-dose leucine-rich essential amino acids stimulates muscle anabolism equivalently to bolus whey protein in older women at rest and after exercise. Am J Phys. 2015;308:E1056–E1065. [PubMed] [Google Scholar]
Churchward-Venne TA, Breen L, Di Donato DM, Hector AJ, Mitchell CJ, Moore DR, Stellingwerff T, Breuille D, Offord EA, Baker SK, Phillips SM. Leucine supplementation of a low-protein mixed macronutrient beverage enhances myofibrillar protein synthesis in young men: a double blind, randomized trial. Am J Clin Nutr. 2014;99:276–286. doi: 10.3945/ajcn.113.068775. [PubMed] [CrossRef] [Google Scholar]
Wilinson DJ, Hossain T, Hill DS, Phillips BE, Crossland H, Williams J, Loughna P, Chruchward-Venne TA, Breen L, Phillips SM, Etheridge T, Rathmacher JA, Smith K, Szewczk NJ, Atherton PJ. Effects of leucine and its metabolite β-hydroxy-β-methlybutryate on human skeletal muscle protein metabolism. J Physiol. 2013;591:2911–2923. doi: 10.1113/jphysiol.2013.253203. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Van Loon LJ. Leucine as a pharmaconutrient in health and disease. Curr Opin Clin Nutr Metab Care. 2012;15:71–77. doi: 10.1097/MCO.0b013e32834d617a. [PubMed] [CrossRef] [Google Scholar]
Ferrando AA, Williams BD, Stuart CA, Lane HW, Wolfe RR. Oral branched chain amino acids decrease whole-body proteolysis. JPEN. 1995;19:47–54. doi: 10.1177/014860719501900147. [PubMed] [CrossRef] [Google Scholar]
Louard RJ, Barrett EJ, Gelfand RA. Effect of infused branched-chain amino acids on muscle and whole body amino acid metabolism in man. Clin Sci. 1990;79:457–466. doi: 10.1042/cs0790457. [PubMed] [CrossRef] [Google Scholar]
Nella 2° parte abbiamo analizzato le caratteristiche e funzioni biochimiche della Glutammina. In questa terza parte, invece, andremo ad analizzare due AA legati tra loro per via metabolica, la L-Citrullina e la L-Arginina.
Dalla L-Citrullina alla L-Arginina – Biologia e principali attività:
Il composto organico Citrullina è un α-amminoacido (formula H2NC(O)NH(CH 2)3CH(NH2)CO2H. ).[1] Sebbene sia stato nominato e descritto dai gastroenterologi fin dalla fine del XIX secolo, è stato isolato per la prima volta dall’anguria nel 1914 dai ricercatori giapponesi Yotaro Koga e Ryo Odake [2] [3] e ulteriormente codificato da Mitsunori Wada dell’Università Imperiale di Tokyo nel 1930.[4] La L-Citrullina è un composto aminoacidico non proteico (non viene utilizzato per formare proteine strutturali come gli enzimi) e, a differenza della L-Arginina, non è ampiamente presente in tutte le proteine. Si trova in concentrazioni particolarmente alte nell’anguria (da cui deriva il suo nome, dato che i cocomeri sono conosciuti come Citrullus vulgaris[1]), dove si trova in media a 2,1mg/g di peso umido (anche se i numeri assoluti variano)[2] e si è notato che il consumo di anguria aumenta in modo acuto sia l’Arginina plasmatica che la Citrullina (3.3 kg di anguria equivalgono a 10g di L-Arginina supplementare)[3][4] e di aumentare l’Arginina e l’Ornitina a digiuno del 12-22% in seguito al consumo di 780-1560g al giorno.[5]
Altre fonti alimentari di L-Citrullina sono i meloni, i meloni amari, le zucchine, le zucche e i cetrioli.[6]
La Citrullina viene sintetizzata nell’organismo attraverso una delle due vie: riciclata dall’Arginina (la conversione dell’arginina in ossido nitrico lascia la citrullina come sottoprodotto)[7][8] o prodotta dall’azoto (e da una parte del carbonio) contenuto nella L-glutammina,[9] dove l’enzima ornitina transcarbamilasi utilizza sia l’Ornitina che il carbamoilfosfato (che richiede la Clutammina) per produrre Citrullina negli enterociti.[10][11]
Sembra che la via dell’Arginina sia responsabile di circa il 10% della Citrullina circolante, mentre la via della Glutammina ne rappresenta il 90%;[6] la riduzione dei livelli plasmatici di Glutammina può ridurre la Citrullina plasmatica.[12]
Per quanto riguarda il ciclo dell’urea (uno dei meccanismi alla base del 10%), la L-Arginina viene convertita in L-Ornitina tramite l’enzima arginasi (cedendo urea come cofattore)[13][14] e da qui l’Ornitina (utilizzando il carbamoilfosfato come cofattore) viene sottoposta all’enzima Ornitina carbamoiltransferasi per produrre L-Citrullina. In questo senso, la via metabolica dall’Arginina alla Citrullina (attraverso l’Ornitina) provoca un aumento dell’urea e una concomitante diminuzione dell’ammoniaca, utilizzata dall’enzima carbamoilfosfato sintasi per creare carbamoilfosfato.[15] Se necessario, l’arginina può essere convertita direttamente in L-Citrullina attraverso un enzima arginina deiminasi per produrre, anziché richiedere, ammoniaca.[16]
Il ciclo si forma quando la citrullina si lega con l’L-aspartato (correlato all’acido D-aspartico come isomero) per formare l’arginosuccinato attraverso l’enzima arginosuccinato sintasi, quindi l’enzima arginosuccinato lisasi degrada l’arginosuccinato in arginina libera e fumarato; l’arginina rientra quindi nel ciclo dell’urea. [Il fumarato può semplicemente entrare nel ciclo TCA (Krebs) come intermedio energetico,[17] e la citrullina regola negativamente l’enzima arginasi.[18]
Anche la conversione della citrullina in L-arginosuccinato e la successiva conversione in L-arginina è coinvolta nel ciclo dell’ossido nitrico piuttosto che nel ciclo dell’urea, con l’unica differenza che l’arginina si converte direttamente in citrullina (cedendo una molecola di ossido nitrico) piuttosto che essere convertita indirettamente tramite l’ornitina.[18][19]
Come accennato, l’Arginina entra prima in contrata con il metabolismo intestinale e splancnico, in cui una certa quantità di essa viene consumata dagli enterociti o interconvertita in L-citrullina o L-ornitina. Oltre all’elevato utilizzo dell’arginina da parte del fegato, anche l’assorbimento intestinale dell’arginina è scarso in condizioni normali e aumenta in varie patologie.[20] Sembra che una quantità minima di L-arginina arrivi ai tessuti sistemici rispetto agli altri aminoacidi del ciclo dell’urea, dato che la L-ornitina supplementare raggiunge una concentrazione sierica doppia rispetto alla L-arginina e la L-citrullina 9,3 volte superiore. Ciò sembra direttamente correlato al grado di metabolismo epatico e intestinale.[21][22][23]
L’Arginina alimentare rappresenta il 40-60% dell’arginina sierica, come evidenziato da un calo equivalente durante i periodi di assenza di arginina. Il tasso di conversione della L-citrullina in L-arginina non sembra influenzato dall’assunzione con la dieta.[24]
La citrullina di per sé è più che altro un sottoprodotto del metabolismo dell’arginina (ciclo dell’ossido nitrico) e dell’ornitina (ciclo dell’urea) e viene semplicemente riconvertita in arginina tramite l’arginosuccinato. Detto questo, l’integrazione di citrullina influisce positivamente anche sulle concentrazioni di arginina e ornitina, quindi anche la loro bioattività è rilevante.
L’arginina può essere convertita in L-citrullina attraverso gli enzimi dell’ossido nitrico sintasi (NOS), di cui esistono forme endoteliali (eNOS) e neuronali specifiche (nNOS), nonché una forma inducibile (iNOS) che risponde ai segnali infiammatori. La conversione dell’arginina attraverso gli enzimi NOS produce ossido nitrico come sottoprodotto più importante, e la Citrullina è vista come un sottoprodotto.[25] La Citrullina può poi riconvertirsi in L-arginina attraverso l’arginosuccinato, ma la L-ornitina non è coinvolta nella via dell’ossido nitrico.
La L-Citrullina viene assorbita nell’intestino in misura molto maggiore rispetto alla sua controparte L-Arginina e determina un livello plasmatico più elevato di L-Arginina attraverso il ciclo Arginina/Ornitina/Citrullina.[26] Viene assorbita attraverso numerosi trasportatori sodio-dipendenti.[27]
È stato osservato che l’integrazione orale di citrullina nell’uomo a 0,18 g/kg raddoppia l’arginina plasmatica[28], cosa che è stata replicata altrove[29], insieme a un aumento equivalente delle concentrazioni di ornitina[29], ma questi raddoppi di arginina e ornitina sono associati a un aumento di 6-11 volte della citrullina plasmatica[28][29].
Una singola dose di 6 g di citrullina malato (0,08 g/kg) in atleti prima dell’esercizio fisico ha fatto registrare aumenti della citrullina plasmatica (aumento del 173%), dell’ornitina (aumento del 152%) e dell’arginina (aumento del 123%) quando misurata dopo l’esercizio fisico, valori che si sono normalizzati con 3 ore di riposo.[30] Questa stessa dose è stata notata altrove per aumentare la citrullina plasmatica e l’arginina in misura simile.[31]
È interessante notare che gli studi sopra citati che hanno utilizzato 0,18 g/kg di citrullina hanno rilevato un aumento di 6-11 volte della citrullina a fronte di un mero raddoppio dell’arginina e dell’ornitina[28][29], mentre lo studio successivo che ha utilizzato 6 g (calcolati come 0,08 g/kg) ha registrato un aumento molto minore della citrullina, ma ha comunque più che raddoppiato sia l’arginina che l’ornitina. [30] Ciò è stato osservato anche in uno studio dose-risposta che ha utilizzato da 2 a 15 g di citrullina, in cui la citrullina nel plasma ha seguito una dipendenza lineare dalla dose, mentre sia l’arginina che l’ornitina hanno avuto una dipendenza minore dalla dose.[29] Gli autori hanno ipotizzato che, dato che l’aumento dell’arginina è stato inferiore a quello previsto e che la citrullina sierica è il principale predittore della sintesi dell’arginina[19], ciò indichi il raggiungimento di una fase di limitazione della velocità nei reni.
È stato osservato che la citrullina non influenza i livelli sierici degli aminoacidi a catena ramificata a riposo,[21] ma può accelerare la deplezione dei BCAA indotta dall’esercizio fisico prolungato (aumentandone l’utilizzo come carburante).[20]
Con l’integrazione di citrullina è stata notata una riduzione della glutammina (13% dopo 0,18 g/kg di citrullina per 7 giorni)[21], anche se un altro studio ha rilevato che l’uso acuto di 6 g di citrullina (0,08 g/kg) non ha alterato le concentrazioni di glutammina.[20]
Gli altri aminoacidi testati (acido glutammico, acido aspartico, asparagina, alanina, lisina, triptofano, fenilalanina, L-tirosina, istidina) sono per lo più inalterati.[20]
Circa l’83% della citrullina ingerita per via orale sembra essere assorbita dai reni[26][27][28] dove viene convertita in L-arginina nei tubuli prossimali (attraverso gli enzimi arginosuccinato sintasi e arginosuccinato liasi[29]); Questa conversione della citrullina in arginina (sia da citrullina supplementare che da quella prodotta come sottoprodotto della creazione di ossido nitrico da parte dell’arginina) rappresenta il 5-15% dell’arginina circolante. [11][30]
Il meccanismo principale con cui l’integrazione di arginina (e, per estensione, di L-citrullina) influisce sulla salute del sangue è quello di essere il substrato per gli enzimi dell’ossido nitrico sintasi (NOS) per la produzione di ossido nitrico, che poi segnala attraverso i recettori ciclici solubili della guanilina la produzione di cGMP. La produzione di ossido nitrico e la successiva produzione di cGMP intracellulare sono alla base di buona parte dei benefici dell’arginina.
Gli enzimi NOS si presentano in tre isoforme principali: [32][33] la NOS inducibile (iNOS), che viene creata in risposta a fattori di stress infiammatori, la NOS neuronale (nNOS), che è stata scoperta per la prima volta nei neuroni e si trova anche nelle terminazioni motorie dei muscoli scheletrici, e la NOS endoteliale (eNOS), che inizialmente si pensava si trovasse solo nell’endotelio, ma è piuttosto diffusa[34], compreso il tessuto cerebrale.[35][36]
Gli enzimi NOS lavorano in dimeri uniti testa a testa e i meccanismi catalitici dipendono da questa dimerizzazione, oltre che dall’eme, dalla tetraidrobiopterina, dalla calmodulina, dal NADPH (come donatore di elettroni) e da FMN e FAD.[37][38][39] Di conseguenza, gli enzimi NOS (tutte e tre le isoforme) sono flavoproteine che richiedono NADPH. [40][41][42] La loro struttura e funzione è complessa (esaminata qui[43]), ma esiste un sito di legame di base per l’arginina e gli elettroni donati dal NADPH fanno sì che l’arginina si converta in citrullina, rilasciando come sottoprodotto l’ossido nitrico; l’iNOS utilizza esclusivamente e l’eNOS può anche utilizzare un intermedio radicale libero chiamato Nω-idrossi-L-arginina (L-NOHA), che si degrada in citrullina e ossido nitrico in presenza di H2O2.[32][44]
L’aumento dell’ossido nitrico (solitamente misurato attraverso le concentrazioni plasmatiche di nitrato/nitrito, citrullina o cGMP urinario) sembra essere aumentato con la L-arginina in persone affette da ipertensione essenziale,[45] arterotrombosi,[46] e diabete di tipo II. [47] Gli studi condotti su atleti altrimenti sani che assumono L-arginina sono piuttosto contrastanti; ci sono casi in cui i biomarcatori del metabolismo dell’ossido nitrico sono aumentati[48] mentre altri studi non notano alcuna modifica.[49][50][51] Non sorprende che i benefici associati all’ossido nitrico non si verifichino quando i biomarcatori dell’ossido nitrico non sono aumentati.
L’inaffidabilità dell’aumento dell’ossido nitrico da parte dell’arginina può essere dovuta al fatto che le concentrazioni fisiologiche di arginina (40-100µM nello spazio extracellulare[52] e forse fino a 800µM a livello intracellulare[53]) sono sufficienti a saturare intrinsecamente l’ossido nitrico sintasi endoteliale (eNOS) (di solito si dichiara una Km di 3µM[54][55], ma a volte viene misurata fino a 29μM[56). Ciò implica che l’enzima è già al massimo dell’efficacia e che un’ulteriore integrazione non aumenta il tasso di conversione (a causa di un arretrato di arginina nel siero); l’osservazione che l’arginina aumenta ancora l’ossido nitrico a volte (anche se in modo inaffidabile) è indicata come il paradosso della L-arginina.[57][58]
Questa teoria è in linea con le osservazioni secondo cui a volte il metabolismo dell’ossido nitrico non viene influenzato nonostante aumenti fino al 300% dell’arginina plasmatica.[59]
Uno studio ha osservato un aumento transitorio della produzione di ossido nitrico che sembra essere più simile a quello di un agonista che di un substrato[60] e successivamente è stato scoperto che l’arginina ha la capacità di attivare i recettori α2-adrenergici,[61] che possono stimolare direttamente l’ossido nitrico senza richiedere la conversione in citrullina attraverso la NOS. Tuttavia, l’arginina è risultata piuttosto debole (superata dall’agmatina)[61] ma questo meccanismo non è ancora stato escluso.
Inoltre, l’arginina extracellulare sembra essere un fattore determinante per il rilascio di ossido nitrico[56] (il trasportatore CAT1 che trasporta l’arginina è altamente associato alla eNOS[62] e l’inibizione dell’afflusso extracellulare impedisce l’attivazione della eNOS[63]), mentre la concentrazione intracellulare di arginina non sembra essere associata. [58] Poiché il trasporto è necessario, ma l’arginina intracellulare non è di per sé necessaria, si ritiene che la colocalizzazione di CAT1 con eNOS[62][64] possa svolgere un ruolo nella stimolazione dell’attività di eNOS.
ADMA
L’ADMA è un metabolita metilato dell’arginina e sembra agire in opposizione all’arginina inibendo le azioni dell’enzima NOS e la conseguente produzione di ossido nitrico. Livelli eccessivi di ADMA possono essere causati da fattori di stress ossidativo che diminuiscono l’attività dell’enzima che lo degrada, mentre la riduzione dell’ADMA provoca una vasodilatazione dovuta alla produzione di ossido nitrico.
Sebbene la maggior parte delle evidenze suggerisca che l’ADMA non aumenta con l’integrazione di L-arginina (questi studi notano che il rapporto arginina:ADMA è aumentato a causa dell’aumento dell’arginina plasmatica), ci sono prove limitate che suggeriscono un aumento che richiede ulteriori indagini.[38]
L’integrazione orale di arginina (anche la citrullina si applica in questo caso perché aumenta l’arginina plasmatica) è in grado di aumentare il flusso sanguigno nelle persone con flusso sanguigno ridotto e, sebbene abbia il potenziale di ridurre la pressione sanguigna, sembra un po’ inaffidabile e può verificarsi solo negli ipertesi. Esistono prove contrastanti sugli effetti dell’integrazione di arginina sul flusso sanguigno in persone con resistenza periferica o cladicazione intermittente, con studi a breve termine che notano un beneficio e studi a più lungo termine che notano un’alterazione.[65][66]
L’integrazione di citrullina sembra ridurre la pressione sanguigna e migliorare il flusso sanguigno in situazioni in cui il flusso sanguigno è altrimenti ostacolato o la pressione sanguigna è più alta del normale, ma la citrullina non ha effetti di riduzione unidirezionali; può essere inefficace in persone normotese a riposo.[41]
In atleti allenati a cui sono stati somministrati 6 g di citrullina malato prima di un test ciclistico prolungato (137 km), l’aumento dell’ormone della crescita indotto dall’esercizio sembra essere aumentato; quando è stato misurato subito dopo l’esercizio, il gruppo con citrullina aveva concentrazioni di GH più elevate del 66,8%, che (dopo 3 ore di riposo) si sono attenuate al 28%.[20] Altrove, dosi di 2-15 g di citrullina non sono riuscite a influenzare l’ormone della crescita a riposo, se misurate nell’arco di 8 ore.[22]
Le concentrazioni di IGF-1 dopo 0,18 g/kg di citrullina per 7 giorni non sono state influenzate in modo significativo.
Durante l’esercizio fisico, sebbene uno studio che ha utilizzato 3 g di L-arginina (associata a 2.200 mg di L-ornitina e 12 mg di vitamina B12) per 3 settimane abbia rilevato un aumento del 35,7% della secrezione di ormone della crescita indotta dall’esercizio fisico (che si è normalizzata entro un’ora)[67], altri studi notano il contrario; è stato osservato che l’integrazione di arginina determina un minore picco di ormone della crescita durante l’esercizio fisico rispetto all’esercizio fisico da solo[68][69] e che, sebbene sembri influenzare maggiormente i giovani rispetto agli anziani[70], si dice che influisca su entrambi i gruppi di età. [L’entità della soppressione (supponendo che il 100% sia il valore di base) è stata notata intorno a un aumento del 300-500% visto con l’esercizio fisico, attenuato al 200%.[68]
È possibile che un aumento eccessivo dell’ormone della crescita stimoli un feedback autogeno, il che spiegherebbe come gli individui più anziani siano meno sensibili a questa soppressione, in quanto hanno intrinsecamente meno picchi di GH dovuti all’esercizio fisico rispetto ai giovani.[71] Infine, poiché i picchi dell’ormone della crescita si normalizzano comunque nel giro di poche ore[67][71], non si sa esattamente quanto sia preoccupante questa soppressione (dato che le concentrazioni di ormone della crescita nelle 24 ore sono più rilevanti).
A riposo, l’integrazione di 5-9 g di L-arginina è in grado di provocare un aumento delle concentrazioni di picco dell’ormone della crescita (aumento del 34,4-120%), mentre 13 g sono risultati inefficaci a causa della sofferenza intestinale che impedisce l’assorbimento della L-arginina.[36]
Negli studi che misurano la secrezione di GH nelle 24 ore, non sono state riscontrate alterazioni significative con la somministrazione di 2 g due volte al giorno[72] o con dosi acute di 5 g.[73] Ciò è potenzialmente legato a un noto fenomeno di feedback autonomo dell’ormone della crescita,[69][74][75] e un effetto modulatorio simile sull’ormone della crescita si riscontra anche durante la restrizione del sonno (in cui una riduzione del rilascio di ormone della crescita indotto dal sonno viene compensato durante le ore di luce). È stato osservato che l’arginina ad alte dosi (250mg/kg di arginina aspartato al giorno, circa 17,5g di arginina) aumenta l’impulso di GH nel sonno a onde lente di circa il 60%, pur non avendo un’influenza sufficiente sulle concentrazioni di GH durante la veglia.[76] Non è chiaro come questo grande aumento influisca sulle concentrazioni di ormone della crescita nell’intera giornata.
L’integrazione di arginina nei ratti è in grado di aumentare il nitrato urinario post-esercizio, indicativo della produzione di ossido nitrico.[77] Aumenti nella produzione di ossido nitrico (nitrato urinario o sierico) sono stati confermati anche nell’uomo in seguito ad assunzione orale o infusione endovenosa.[78]
Non sempre si riscontra un aumento della produzione di ossido nitrico (anche nonostante l’aumento dell’arginina plasmatica), suggerendo che l’attività dell’enzima NOS potrebbe essere un fattore limitante. Per quanto riguarda gli studi in acuto (assunzione di una singola dose di L-arginina prima dell’esercizio), 3 g di arginina (sotto forma di AAKG) non hanno apportato benefici all’allenamento con i pesi,[79] 6 g di L-arginina per 3 giorni non hanno modificato i risultati del cicloergometro in atleti di judo, mentre un protocollo simile in ciclisti allenati ha rilevato un miglioramento del tempo di esaurimento (25,8%).[80]
Alcuni studi hanno utilizzato una forma di arginina nota come GAKIC (Glycine L-Arginine α-Ketoisocaproic acid) e hanno rilevato un aumento della potenza media durante gli sprint di 10s su cicloergometro (con 11,2 g di GAKIC)[81] e un aumento del 10,5+/-0. Questi studi, tuttavia, sono confusi sia dall’inclusione della glicina sia da quella del metabolita della leucina, l’acido α-chetoisocaproico.
Per quanto riguarda gli studi più cronici, l’integrazione di L-arginina (come asparato) con 2,8 o 5,7 g di arginina al giorno per 4 settimane non è riuscita a modificare le prestazioni o altri biomarcatori[82]; anche uno studio precedente, condotto per 2 settimane con una metodologia simile, ha fallito.[83] Nel complesso, quando si esaminano le revisioni o le meta-analisi sull’argomento L-arginina e prestazioni sportive, si nota che è promettente, ma manca un consenso sufficiente per raccomandarla come ergogenico.[84]
Si ritiene che la citrullina sia un agente pro-erettile in quanto è un precursore dell’arginina, e l’arginina è il substrato da cui viene prodotto l’ossido nitrico che può poi indurre il cGMP (attraverso la via NO/cGMP/VEGF);[65] un aumento del cGMP è anche l’effetto finale degli inibitori della PDE5 come il viagra o l’icariina dall’erba cornuta.[66]
Negli uomini con disfunzione erettile, valutata come debolezza dell’erezione (valutata con il punteggio di durezza yerettile[67]), la somministrazione di 1.500 mg di citrullina al giorno (due dosi da 750 mg) per un mese è stata in grado di apportare benefici alla metà dei 24 pazienti valutati (valutati come “molto soddisfatti” del trattamento), mentre il miglioramento del placebo è stato solo dell’8,3%.[68]
La citrullina sembra interagire con il metabolismo dei BCAA nell’organismo, anche se gli studi sull’uomo sembrano avere risultati diversi a seconda del contesto dello studio.
La citrullina può mediare positivamente la segnalazione della leucina attraverso mTOR, il che suggerisce teoricamente una sinergia. L’applicazione di questa combinazione ai sollevatori di pesi non è ancora stata studiata, quindi il sinergismo è attualmente solo un’ipotesi piuttosto che un fatto dimostrato.
Il nitrato è un piccolo donatore di ossido nitrico che costituisce il principale bioattivo del succo di barbabietola. Il nitrito sierico (forma ridotta del nitrato) sembra aumentare durante l’esercizio fisico in seguito al consumo di 6 g di citrullina malato, che si ritiene sia un indicatore dell’aumento della produzione di ossido nitrico.[20]
I farmaci a base di statine possono aumentare l’espressione dell’enzima che media la conversione dell’arginina in ossido nitrico e per questo motivo è possibile che vi sia un sinergismo per tutto ciò che riguarda l’ossido nitrico. Questo non è ancora stato testato in un sistema vivente.
L-Citrullina come sostituto alla L-Arginina?
L’integrazione di L-citrullina è stata definita un’alternativa alla L-arginina, in quanto ne aggira lo scarso assorbimento e si converte in L-arginina nei reni. La L-citrullina tecnicamente segue aumenti dose-dipendenti della L-arginina sierica fino a 15 g, ma la dose orale più alta di citrullina assunta ha ritorni sempre minori (cioè per ogni 5 g in più di citrullina ingerita si aggiunge meno arginina al siero).[85]
È stato osservato che la citrullina orale a 0,18 g/kg raddoppia approssimativamente l’arginina plasmatica (aumento del 100%)[86][87] o è leggermente superiore (123%) con 0,08 g/kg.[88] Poiché le dosi più elevate di L-citrullina hanno una minore conversione in arginina[85], è improbabile che la L-citrullina supplementare possa essere utilizzata per superare l’arginina per l’aumento acuto dell’arginina sierica.
Gli studi che hanno confrontato direttamente la L-arginina con la L-citrullina hanno osservato che entrambe aumentano la Cmax a livelli comparabili a dosi orali simili (Cmax di 79+/-8μM per 3 g di citrullina e 84+/-9μM per l’arginina), ma la citrullina risulta in un’AUC complessiva maggiore (48,7% in più rispetto all’arginina). [Questo potrebbe essere dovuto al fatto che, anche fino all’ingestione di 15g di citrullina, non si verifica un aumento significativo dell’escrezione di citrullina.[85] L’assenza di un aumento dell’eliminazione di L-citrullina dal sangue nonostante l’integrazione consentirebbe di avere un pool di L-citrullina disponibile per la conversione su richiesta in L-arginina.
Citrullina Malato:
La Citrullina Malato (CM), una combinazione di L-citrullina e acido malico, è stata pubblicizzata come un aiuto ergogenico (che aumenta l’energia) per l’allenamento contro-resistenza e l’esercizio ad alta intensità.
Come abbiamo visto, la L-citrullina è un precursore dell’ossido nitrico (NO), un vasodilatatore che può migliorare l’apporto di sangue e ossigeno ai muscoli durante l’esercizio. Tuttavia, le prove finora disponibili suggeriscono che il miglioramento del flusso sanguigno non è il meccanismo attivo degli effetti ergogenici del CM. Il meccanismo potrebbe invece essere dovuto alla capacità della citrullina di favorire l’eliminazione dell’ammoniaca durante l’esercizio ad alta intensità, alla capacità del malato di aumentare la produzione di ATP, a un aumento dell’espressione genica o a una maggiore efficienza della navetta malato-aspartato.
La maggior parte delle ricerche condotte finora ha utilizzato una dose acuta di 8 grammi di CM un’ora prima dell’esercizio. Sebbene l’assunzione di CM un’ora prima dell’esercizio rimanga la migliore raccomandazione, alcuni dati suggeriscono che dosi maggiori, fino a 15 grammi, potrebbero essere più benefiche.
È stato dimostrato che l’ingestione di una serie di dosi di CM (2-15 grammi) non ha effetti negativi sui marker ematologici. Sebbene la sicurezza di un’integrazione di CM a lungo termine richieda ulteriori indagini, le ricerche condotte finora indicano che la CM è ben tollerata nella maggior parte degli individui.
Ricerche preliminari hanno suggerito che 8 grammi di CM ingeriti un’ora prima dell’esercizio fisico aumentano la resistenza muscolare (ripetizioni fino al cedimento) in uomini e donne. Tuttavia, ricerche più recenti hanno suggerito che il CM potrebbe non avere un beneficio sulle prestazioni nell’allenamento contro-resistenza, potenzialmente a causa di variazioni nei tempi e nei dosaggi.[89]
Arginina AKG:
La differenza principale tra la L-arginina e Arginina AKG è che la L-arginina è un aminoacido non essenziale che l’organismo è in grado di produrre, mentre l’arginina AKG è un sale della L-arginina e dell’acido α-chetoglutarato. Inoltre, la L-arginina regola il flusso sanguigno attraverso la produzione di ossido nitrico, mentre l’Arginina AKG dovrebbe potenzialmente aumentare il flusso sanguigno, l’energia e il recupero.
Nella nutrizione sportiva, l’AKG è stato utilizzato come integratore per migliorare la sintesi proteica muscolare e diminuire la disgregazione muscolare, ed è quindi utilizzato dagli atleti per migliorare la composizione corporea.[90][91] L’integrazione di AKG potrebbe anche migliorare le prestazioni atletiche. Uno studio ha rilevato che un integratore di arginina e alfa-chetoglutarato (AAKG) ha migliorato la forza nella panca, ma non la capacità aerobica. Sono necessarie ulteriori ricerche per sostenere le affermazioni sull’AKG come aiuto ergogenico.[92]
L’AKG viene utilizzato anche per il recupero da interventi chirurgici o traumi, perché è un precursore dell’aminoacido glutammina. Sebbene la glutammina sia un aminoacido non essenziale, viene talvolta definita “condizionatamente essenziale” perché la quantità di glutammina di cui l’organismo ha bisogno per il recupero dopo un trauma significativo può superare la quantità che l’organismo è in grado di produrre. In questo caso, un integratore di AKG può aiutare il processo di recupero.[93][94]
L’AKG è stato proposto come integratore per la longevità; alcune ricerche condotte su vermi tondi, ratti e topi suggeriscono che potrebbe aumentare la durata della vita e ritardare l’insorgenza di malattie legate all’età, anche se gli studi clinici dovranno confermare questi risultati.[95][96]
Nelle persone con malattie renali croniche, in particolare in quelle sottoposte a dialisi, la somministrazione di AKG in combinazione con il calcio ha migliorato i biomarcatori della funzione renale.[97][98]
In uno studio è stato rilevato che l’AKG aumenta l’espressione di involucrina, filaggrina e serina palmitoil transferasi. Queste molecole sono tutte importanti per la struttura dello strato esterno della pelle e per l’idratazione dello strato esterno della pelle, quindi l’uso di AKG per via topica potrebbe migliorare l’aspetto della pelle.[99][100]
Nella ricerca, i dosaggi utilizzati variano da 3,6 g a 6 g, con dosaggi più elevati nelle persone che hanno subito ustioni, ma non è ancora stata stabilita una dose giornaliera raccomandata.[101] Poiché gli effetti sono dose-dipendenti, trovare una raccomandazione di dosaggio accurata sarà una parte importante della ricerca in corso.[102]
Sicurezza e tossicità:
La citrullina sembra essere ben tollerata dai ratti in dosi fino a 3 g/kg di peso corporeo[58][46]. Negli esseri umani, 15 g di citrullina assunti acutamente non sembrano causare diarrea o disturbi intestinali[22], il che è notevolmente diverso rispetto all’ornitina e all’arginina che possono causare diarrea a dosaggi di 10 g se assunti in bolo[74][75] a causa del limitato assorbimento di questi aminoacidi che poi procedono verso il colon causando diarrea osmotica.[74]
Il limite di sicurezza osservato, ovvero la dose più alta in cui si può essere relativamente sicuri che non si verifichino effetti collaterali nel corso della vita, è stato suggerito in 20 g di arginina al giorno in forma di integratore (al di sopra dell’assunzione di cibo).[103] Dosi più elevate sono state testate e ben tollerate, ma non esistono prove che suggeriscano la loro sicurezza in tutte le popolazioni nel corso della vita.
La L-arginina ha un tasso di assorbimento gastrointestinale piuttosto scarso. Può inoltre agire come assorbente, rilasciando acqua ed elettroliti nel lume intestinale attraverso la stimolazione dell’ossido nitrico e inducendo disturbi gastrici e diarrea.[12] Questo fenomeno è noto come diarrea osmolitica e tende a verificarsi a dosi orali superiori a 10 g circa, se assunte in bolo.[36]
Si pensa che ciò avvenga attraverso la stimolazione della produzione di ossido nitrico, poiché la D-Arginina (incapace di produrre NO) non produce diarrea[104] e l’ossido nitrico stesso è noto per essere un meccanismo attraverso il quale molti lassativi osmolitici funzionano.[105]
Singoli boli di 5-9 g di L-arginina senza cibo non sembrano causare disturbi intestinali come dosi superiori a 10 g,[36] suggerendo che, almeno per uno stomaco vuoto, il dosaggio di 9 g è un limite superiore.
Conclusioni:
L’Integrazione di L-citrullina si è dimostrata più redditizia tra costi e benefici (vedi assorbimento intestinale) rispetto alla L-arginina. L’uso alternativo di Citrullina Malato può portare ad eventi gastrointestinali più frequenti rispetto alla semplice forma L-citrullina. Nonostante la ridotta biodisponibilità orale della L-arginina, questa può essere mixata con L-citrullina per un effetto additivo, anche se non rappresenta un vero e proprio vantaggio proprio di tale abbinamento.
L’assunzione di L-arginina e/o L-citrullina vede la sua miglior tempistica prima dell’allenamento al fine di aumentare il flusso sanguigno ai distretti allenati, per effetto della vasodilatazione NO indotta.
Ciò si traduce in:
Aumento dell’apporto di nutrienti e ossigeno al tessuto muscolare abbinato ad un effetto di pulizia dalle molecole di scarto, come l’acido lattico;
Esaltazione dilatatoria sul reticolo venoso sottocutaneo, che migliora la qualità estetica in definizione.
Bonus: l’abbinamento con estratto di barbabietola notoriamente ricco di nitrati.
Come effetto diretto dell’introduzione nell’organismo di estratto di barbabietola abbiamo un aumento della sintesi di ossido nitrico (NOs), dovuta, per l’appunto, ai nitrati (NO3-) contenuti in questo vegetale, convertiti rapidamente in nitriti (NO2-2) tramite enzimi che si trovano fin dal tratto orofaringeo, gastrointestinale e tracheo-bronchiale. Dato ciò, la sintesi di NO sfrutta un percorso non usuale come quello della L-Arginina, ma coadiuvante a questa e alla L-Citrullina.
Le concentrazioni di nitrati raggiungono il picco dopo circa un’ora dalla sua ingestione, per ritornare ai livelli basali dopo quasi 24h, mentre gli effetti della L-Arginina permangono per almeno 75-80 minuti, per poi iniziare a tornare ai livelli basali.
“Citrulline – Compound Summary”. PubChem Compound. USA: National Center for Biotechnology Information. 16 September 2004. Identification. Retrieved 1 May 2012.
Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, Jung G, Solis GM, Fazlollahi F, Kaweeteerawat C, Quach A, Nili M, Krall AS, Godwin HA, Chang HR, Faull KF, Guo F, Jiang M, Trauger SA, Saghatelian A, Braas D, Christofk HR, Clarke CF, Teitell MA, Petrascheck M, Reue K, Jung ME, Frand AR, Huang JThe metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR.Nature.(2014-Jun-19)