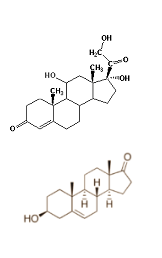
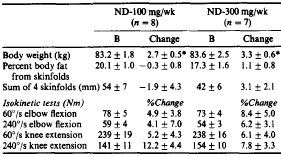L’Exemestane (Aromasin) è un farmaco inibitore della aromatasi di terza generazione, come l’Anastrozolo (Arimidex) e il Letrozolo (Femara), utilizzato nel trattamento del carcinoma della mammella. L’ Anastrozolo e il Letrozolo sono molto efficaci nell’inibizione della conversione degli Androgeni in Estrogeni. Il Letrozolo, per esempio, presenta una capacità di abbassare gli Estrogeni del 98% e oltre mentre l’Anastrozolo del 70%. Questi due farmaci hanno un largo uso in ambito sportivo rispetto all’Exemestane , ma a differenza di quest’ultimo sono inibitori reversibili e ciò vuol dire che essi si legano all’aromatasi senza distruggerlo. Ciò può portare a rebound estrogenici dopo l’interruzione dell’uso di questi farmaci. L’Exemestane, invece, è un inibitore inreversibile dal momento che legandosi all’enzima aromatase lo distrugge, evitando così la possibile comparsa di rebound una volta interrotto il suo utilizzo. Nonostante l’uso del Letrozolo può sembrare superiore, con il suo potente effetto di riduzione estrogenica, bisogna ricordare che gli estrogeni, tra le altre cose, sono necessari per una salute ottimale delle articolazioni e per un sistema immunitario sano ed efficiente. Così abbassarli del 98% per un periodo prolungato di tempo, non può essere la migliore delle idee. Questo può essere utile in un ciclo pre-gara di BodyBuilding, o se si è particolarmente soggetti a sviluppare ginecomastia, ma certamente non risulterebbe utile e sicuro per lunghi periodi di tempo, senza compromettere le articolazioni e il sistema immunitario (senza dimenticarci del rebound).
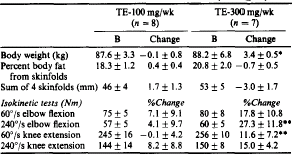
L’Arimidex, che non è così potente come il Letrozolo, a 0.5 mg/giorno è in grado di abbassare gli estrogeni del 50%. Ciò rende l’Arimidex una buona scelta durante i cicli, se non si è molto inclini a sviluppare ginecomastia o se non ci si sta preparando per una gara.
Ma per quanto riguarda la PCT?
Credo che a questo punto la maggior parte delle persone propenderà per l’uso del Nolvadex (Tamoxifen Citrato) invece del Clomid per la terapia post ciclo (PCT), anche se entrambi competono per il sito recettore degli estrogeni, aumentano i livelli sierici di Testosterone, e entrambi i farmaci possono anche alterare positivamente il profilo lipidico nel sangue. Ma dal momento che 20 mg di Tamoxifene sono pari a 150 mg di Clomifene ai fini dell’aumento dei livelli di Testosterone, dell’FSH e LH, e il Tamoxifene non diminuisce la risposta dell’LH LHRH penso che molte persone propendano per la superiorità del Nolvadex in PCT. In realtà, l’uso combinato del Nolvadex con il Clomid risulta superiore dal momento che il Clomifene è un agonista/antagonista misto (SERM) a livello del recettore dell’Estradiolo. Il Clomifene aumenta la secrezione di LH con un’azione ipotalamo-ipofisi. L’uso del Clomifene causerà un aumento dell’LH e secondariamente un aumento del Testosterone e dell’Estradiolo. L’Estradiolo influisce negativamente sul feedback dell’HPTA. L’Estradiolo ha un effetto inibitorio marcato sul Testosterone. Il Tamoxifene contrasterà l’effetto dell’Estradiolo.
Ho spesso trovato molto utile l’uso del Nolvadex e del Clomid durante una PCT, insieme con un AI; anche perché la riduzione dei livelli di estrogeni esercitata dagli AI è stata positivamente correlata con un aumento del Testosterone . A questo punto ci si può domandare: quale AI usare? Letrozolo o Anastrozolo? Nessuno dei due! Purtroppo, il Nolvadex ridurrà in modo significativo i livelli plasmatici sia del Letrozolo che dell’Anastrozolo. Quindi, se si sceglie di utilizzare uno di questi composti con il Nolvadex in PCT, si getteranno via soldi dato che il Nolvadex ridurrà la loro efficacia.
Ed è in questo particolare momento che l’Aromasin diventa la scelta migliore a 25 mg/giorno.
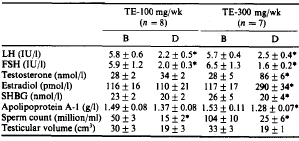
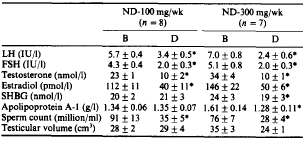
L’Aromasin, a questa dose, oltre a promuovere un aumento dei livelli di Testosterone di circa il 60% e inibire l’aromatizzazione di quest’ultimo, aiuterà il miglioramento del rapporto testosterone libero/testosterone legato abbassando i livelli della globulina legante l’ormone sessuale (SHBG), di circa il 20%.
Per capire il perché l’Aromasin può essere utile in congiunzione con il Nolvadex mentre sia il Letrozolo che l’Arimidex soffrono di ridotta efficacia, dovremo prima capire le differenze tra inibitori dell’aromatasi di tipo I e di tipo II. Gli inibitori di Tipo I (come l’Aromasin) sono in realtà composti steroidei, mentre gli inibitori di Tipo II (come il Letrozolo e l’Arimidex) sono farmaci non steroidei. Quindi, gli effetti collaterali androgeni sono possibile con gli inibitori di tipo I, e dovrebbero essere evitati dalle donne. Gli inibitori di tipo 1, o inattivatori enzimatici steroidei, sono steroidi analoghi dell’androstenedione, che si legano irreversibilmente al medesimo sito della molecola dell’aromatasi; gli inibitori di tipo 2 , o inibitori enzimatici non steroidei, sono sostanze a struttura non steroidea, che si legano reversibilmente al gruppo eme dell’enzima aromatasi. Nel caso di un AI di tipo-I, l’inibitore non competitivo si legherà, e l’enzima avvierà una sequenza di idrossilazione; questa idrossilazione produrrà un legame covalente indissolubile tra l’inibitore e la proteina enzimatica. Ora, l’attività enzimatica viene bloccata in modo permanente; anche se l’assunzione dell’inibitore viene interrotta. L’attività enzimatica dell’aromatasi può essere ripristinata solo da una nuova sintesi degli enzimi. Invece, gli inibitori competitivi, AI di tipo-II, si legano reversibilmente al sito attivo dell’enzima; a questo punto possono accadere due cose: 1) nessuna attività enzimatica viene attivata o 2) l’enzima è in qualche modo attivato senza alcun effetto. L’inibitore di tipo II può effettivamente dissociarsi dal sito di legame, permettendo una rinnovata concorrenza tra l’inibitore e il substrato per il legame al sito. Ciò significa che l’efficacia degli inibitori competitivi dipende dalle concentrazioni relative e dalle affinità sia dell’inibitore che del substrato, mentre questo non avviene per gli inibitori non competitivi. L’Aromasin è un inibitore di tipo I, il che significa che una volta che la molecola ha fatto il suo lavoro, e disattivato l’enzima aromatasi, non si necessita di mantenerne l’assunzione. Il Letrozolo e l’Arimidex necessitano effettivamente di una continua assunzione per mantenere l’effetto. Questo è forse il motivo per cui il Nolvadex non altera la farmacocinetica dell’Aromasin.
L’utilità dell’Aromasin vede un suo possibile utilizzo nella PCT scientifica del dott. Scally da me già esposta. Insieme all’uso di Nolvadex, Clomid e HCG, l’Aromasin darebbe quel tocco aggiuntivo che perfezionerebbe ulteriormente la già logica applicazione dei tre farmaci prima citati. Infatti, l’azione inibitoria dell’Aromasin può essere sfruttata maggiormente nei primi 20 giorni, quando l’uso di HCG a giorni alterni aumenta di conseguenza i livelli di Testosterone e la loro possibile aromatizzazione in estrogeni.
Esempio:
Giorno 1-20: 2000UI di HCG a giorni alterni
Giorno 1-30: 100mg di Clomid al giorno
Giorno 1-45: 40mg di Nolvadex al giorno
Giorno 1-45: 25mg di Aromasin a giorni alterni (emivita 27 ore)
Prima di chiudere il discorso sull’Aromasin, vale la pena notare che si può (e si deve) comunque usare uno degli AI non-steroidei durante un ciclo contenente forti aromatizzabili per ridurre gli estrogeni in eccesso, se necessario. Quando si è pronti per la PCT, è possibile passare all’ Aromasin e continuare a sperimentare gli effetti già mantenuti con i precedenti AI, poiché non vi è alcun cross-over di tolleranza sperimentata tra AI steroidei e non steroidei. Poiché l’Aromasin mediamente esercita una soppressione degli estrogeni del 65%, è certamente un ottimo agente anti aromatase, soprattutto se si considera il fatto che non si verificherà una riduzione dell’efficacia a causa dell’uso concomitante del Nolvadex. Non vi sarà alcun tipo di tolleranza sviluppata utilizzando altri AI. C’è anche una discreta quantità di dati preclinici i quali suggeriscono che l’Aromasin ha un effetto benefico sul metabolismo minerale osseo che non si verifica con gli agenti non steroidei. Può anche avere effetti benefici sul metabolismo dei lipidi, cosa che non si verifica con l’uso di AI non steroidei come il Letrozolo e l’Arimidex.
Gabriel Bellizzi
Riferimenti:
- Clin Cancer Res. 2005 Apr 15;11(8):2809-21.
2. J Clin Endocrinol Metab. 1995 Sep;80(9):2658-60.
[Clinical aspects of estrogen and bone metabolism] Clin Calcium. 2002 Sep;12(9):1246-51. Japanese. - Science, Vol 283, Issue 5406, 1277-1278 , 26 February 1999
J Clin Endocrinol Metab 2000 Jul;85(7):2370-7, “Estrogen Suppression in Males”
Fertil Steril. 1978 Mar;29(3):320-7 - J Clin Endocrinol Metab. 2004 Mar;89(3):1174-80
- .J Steroid Biochem Mol Biol. 2001 Dec;79(1-5):85-91.
- The Oncologist, Vol. 9, No. 2, 126–136, April 2004
- Zilembo N., Noberasco C., Bajetta E., Martinetti A., Mariani L., Orefici S.
- Endocrinological and clinical evaluation of exemestane, a new steroidal aromatase inhibitor. Br. J. Cancer, 72: 1007-1012, 1995
- Clinical Cancer Research Vol. 10, 1943-1948, March 2004
- The Journal of Clinical Endocrinology & Metabolism Vol. 88, No. 12 5951-5956.