Androgeno: 44-57
Anabolico: 88
Standard: Testosterone
Nome chimico: 17beta-Hydroxy-1-methyl-5alpha-androst-1-en-3-one, 1-methyl-1(5-alpha)-androsen-3-one-17b-ol.
Attività estrogenica: nessuna
Attività progestinica: non ci sono dati disponibili (bassa o nulla)
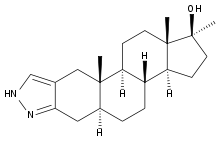
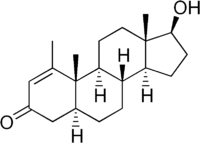
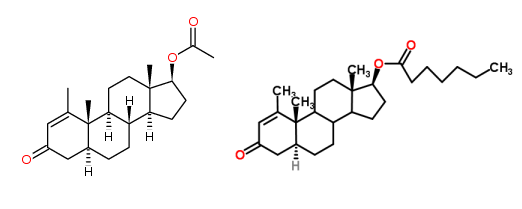
Il Metenolone [17beta-Hydroxy-1-methyl-5alpha-androst-1-en-3-one], o methenolone, noto anche come methylandrostenolone, conosciuto al grande pubblico con il suo vecchio nome commerciale Primobolan (oggi Rimobolan), è un AAS derivato del DHT con valore androgeno/anabolizzante pari a 44-57:88 . Si tratta di una molecola naturale, che si trova all’interno delle ghiandole surrenali dei felini domestici in gravidanza (1), ed è sintetizzata in laboratorio legata all’estere acetato per la somministrazione orale e all’ estere enantato per iniezione intramuscolare.
Il Metenolone è stato descritto per la prima volta nel 1960.(2) Squibb introdusse il farmaco (nella forma orale e iniettabile) negli Stati Uniti nel 1962.(3) Questa molecola è stata venduta per un lasso di tempo molto breve sia nella sua forma orale (in compresse da 20 mg) sia nella sua forma iniettabile, rispettivamente con il marchio di Nibal® e Nibal® Depot . La Schering nella Germania occidentale (ora Bayer) ottenne i diritti sul farmaco che nello stesso anno, avrebbe venduto sotto il nome di Primobolan. Il Nibal® (in entrambe le sue forme) fu presto rimosso dal mercato degli Stati Uniti senza più ritornarvici. Così la Schering ottenne i diritti di brevetto esclusivo per la produzione del Metenolone acetato e enantato, e avrebbe continuato a vendere il farmaco senza interruzioni dal 1962, e per i consumatori divenne naturale identificare il Metenolone come un prodotto Schering.
Così il Primobolan è sempre stato identificata come uno steroide europeo, e nel corso degli anni ’60 e ’70 è stato venduto in paesi come Germania, Austria, Belgio, Francia, Paesi Bassi e Finlandia. Per un periodo di tempo la Schering produsse una forma iniettabile a base oleosa contenente 20mg/ml di Metenolone acetato (chiamato Primobolan Acetato), ma venne dichiarato fuori produzione dal 1993. Il Metenolone acetato iniettabili dimostrò di essere molto popolare per la preparazione alla gara, tanto che quando venne eliminato dal mercato gli atleti europei ne sentirono la mancanza. Anche se continuava ad esistere la forma orale del Metenolone acetato non è uguale (ovviamente la versione iniettabile è molto più efficiente per questo steroide, ma il perché lo vedremo più avanti).
La Schering mantenne il controllo del brevetto del Metenolone fino alla fine del 1970. Prima che i suoi brevetti scadessero, Schering aveva rigorosamente protetto i propri diritti di proprietà intellettuale contro ogni potenziale violazione, anche nel mercato statunitense, dove l’azienda non distribuiva Primobolan. Anche se il Metenolone non è stato più disponibile per la vendita commerciale negli Stati Uniti per decenni, è tecnicamente mantenuto il suo status di farmaco approvato dalla FDA.
Il Primobolan è in genere prescritto come un agente anabolizzante per la crescita del tessuto magro, spesso utilizzato nei casi post-operatori, di infezioni prolungate, di malattie con una forte componente catabolica, in seguito alla somministrazione cronica di corticosteroidi, o in convalescenza. Alcuni medici prescrivono questa molecola per il trattamento dell’osteoporosi, della sarcopenia (la naturale perdita di massa muscolare con l’invecchiamento), alcuni casi di epatite cronica, ed il carcinoma della mammella (di solito come un farmaco secondario seguito da altre terapie). Lo steroide è stato anche utilizzato per promuovere l’aumento di peso nei neonati prematuri sottopeso e nei bambini in studi clinici, ed è stato in grado di farlo in modo efficace e senza segni di tossicità o effetti indesiderabili.(4) Gli atleti hanno a lungo favorito l’uso di questo AAS, con il suo valore androgeno contenuto e la sua natura non-estrogenica, caratteristiche che lo rendono molto favorevole per atleti principianti.
Anche se il Primobolan ha dimostrato un buon margine di sicurezza clinica, dagli anni ’90 la Schering era cresciuta fino a diventare un gigantesca multinazionale farmaceutica, ed è stata inevitabilmente costretta a riesaminare la propria offerta di steroidi a livello mondiale alla luce delle preoccupazioni del pubblico circa il doping nello sport. Il Primobolan sarebbe stato ritirato volontariamente dalla maggior parte dei paesi che lo avevano originariamente venduto. Oggi il Primobolan è venduto in una manciata di paesi, tra cui Spagna, Turchia, Giappone, Paraguay e Ecuador. Nonostante la sua offerta limitata, la Bayer è rimasta (quasi) il produttore esclusivo del Metenolone nel business farmaceutico umano mondiale.
La Schering aveva ritirato la forma orale del farmaco dalla maggior parte dei mercati nei primi anni 2000. Non ci sono tutt’ora versioni orali contenenti 50mg ancora in produzione, ne esistono al massimo un paio di prodotti contenenti 5 mg o 25 mg che possono essere ancora legalmente in circolazione. Le sole fonti confermate per il Primobolan orale in questi ultimi anni sono state in Giappone e in Sud Africa, e questi sono stati venduti sotto il marchio Schering. Non è noto se tali prodotti siano stati venduti anche con la nuova etichetta Bayer. Oltre a questo, un piccolo numero di preparazioni farmaceutiche contenenti Metenolone acetato potrebbero essere ancora in produzione. Negli ultimi anni, tuttavia, il Metenolone è prodotto e distribuito anche dalle UGL del mercato nero.
Come precedentemente accennato, il Metenolone è un derivato del DHT con due modifiche strutturali, e cioè:
- il doppio legame in C1-C2, che incrementa la stabilità del chetone in posizione C3 (fondamentale per il mantenimento del legame con i recettori androgeni muscolari, dove infatti il Metenolone viene degradato a diol; tipico del DHT che subisce un’idrossilazione nelle cellule muscolari quasi istantanea) solo in parte minore; inoltre questa modificazione rende la molecola meno affine al legame con l’SHBG, aumentandone la bio attività.
- il metile in C1 che aumenta in qualche misura la resistenza della molecola al passaggio epatico; non a livello di una metilazione in C17, ma nemmeno tossica come queste. Questa caratteristica ne consente l’utilizzazione anche orale della forma acetata, sebbene a dosaggi molto più alti della forma enantata, tipicamente somministrata per via iniettabile. La presenza del metile in C-1 è l’unica differenza rispetto al Diidroboldenone: essendo quets’ ultimo circa tre volte più potente come anabolizzante rispetto al Metenolone, si presume che questa modificazione della struttura molecolare, anche se aumenta la biodisponibilità orale, ne diminuisca in qualche modo la potenza anabolica, molto probabilmente aumentando l’affinità per l’SHBG, particolarmente elevata nel Metenolone. In pratica la metilazione in C-1, parzialmente inverte l’effetto del doppio legame in C1-C2, rendendo più biodisponibile oralmente il Metenolone rispetto al Boldenone, ma rendendo il primo composto meno bioattivo.
Il Metenolone, essendo una molecola 5-alfa ridotta (in quanto derivato del DHT) non subisce alcuna aromatizzazione (5) e non presenta alcuna attività estrogenica misurabile. Quindi, effetti collaterali estrogeno-dipendenti non dovrebbero essere considerati quando si somministra questo steroide da solo. Anche gli individui sensibili non devono preoccuparsi di sviluppare ginecomastia, o ritenzione idrica apprezzabile con questo farmaco.
Anche se questo steroide presenta un attività androgena pari a 44-57, la comparsa degli effetti collaterali androgeni è ancora possibili. Questo può includere pelle oleosa, acne, crescita di peli su corpo e viso. Anche questo AAS può aggravare la alopecia androgenetica la dove geneticamente predisposti. Le donne devono essere avvertite dei potenziali effetti virilizzanti di questo steroide. Questi possono includere voce profonda, irregolarità mestruale, cambiamenti nella struttura della pelle, crescita di peli sul viso (irsutismo), e l’allargamento del clitoride. Quindi, nonostante il Metenolone sia un AAS molto mite, può presentare forti effetti collaterali androgeni a dosi elevate.
Il Metenolone, non essendo una molecola metilata in C-17, non è considerato uno steroide epatotossico; la tossicità epatica è improbabile. Durante gli studi non si sono osservati cambiamenti apprezzabili nei marcatori dello stress epatico quando il farmaco è stato somministrato a dosi terapeutiche. (5) La forma orale del Metenolone ha una certa resistenza alla de-attivazione epatica, e la tossicità epatica, l’insufficienza, e la morte sono state riportate in un paziente anziano che riceve metenolone acetato orale.(6) Sebbene sia improbabile, l’epatotossicità non può essere completamente esclusa, soprattutto con dosi molto elevate per via orale.
L’assenza della metilazione in posizione C-17 conferisce al Metenolone una capacità tutto sommato lieve di influire sull’equilibrio lipidico. Gli AAS possono avere effetti deleteri sul colesterolo sierico. Questo include una tendenza alla riduzione del colesterolo HDL (buono) e all’aumento del colesterolo LDL (cattivo), con un alterazione dell’equilibrio tra HDL/LDL favorendo un maggiore rischio di arteriosclerosi. L’impatto relativo di un AAS sui lipidi sierici dipende dalla dose, dalla via di somministrazione (per via orale o iniettabile), dal tipo di steroide (aromatizzabile o non aromatizzabile), e dal livello di resistenza al metabolismo epatico. Il Metenolone dovrebbe avere un effetto negativo più forte sulla gestione epatica del colesterolo rispetto al Testosterone o al Nandrolone a causa della sua natura non aromatizzabile, ma molto più debole rispetto agli AAS c-17 alfa alchilati. A causa del metodo di somministrazione, il Metenolone orale presenta un effetto negativo leggermente più forte sui lipidi rispetto alla forma iniettabile. Gli AAS possono anche influenzare negativamente la pressione del sangue e i livelli dei trigliceridi, riducendo il rilassamento endoteliale, e promuovendo l’ipertrofia ventricolare sinistra, tutti fattori con un potenziale nel aumentare il rischio di malattie cardiovascolari e infarto del miocardio.
Per contribuire a ridurre lo sforzo cardiovascolare si consiglia di mantenere un programma di esercizio cardiovascolare attivo e di ridurre al minimo l’assunzione di grassi saturi, colesterolo e carboidrati semplici in ogni momento durante la somministrazione di AAS.
La supplementazione con oli di pesce (4 grammi al giorno) e un integratore alimentare di Niacina per il controllo del colesterolo è anche raccomandata.
Tutti gli AAS se assunti in dosi sufficienti per promuovere l’aumento della massa muscolare causano una soppressione del Testosterone endogeno. Senza l’intervento con sostanze Testosterone-stimolante, e una adeguata PCT, i livelli di Testosterone dovrebbero tornare alla normalità entro 1-4 mesi dalla cessione del farmaco. Si noti che un ipogonadismo ipogonadotropo prolungato può svilupparsi secondariamente all’abuso di steroidi, cosa che richiede un intervento medico. Il Metenolone è generalmente descritto come avente un basso impatto sulla produzione di Testosterone endogeno. Anche se questo può essere vero con piccole dosi cliniche (20-25 mg al giorno), in ambito sportivo, e quindi dopante, le cose sono diverse. In uno studio, più della metà dei pazienti trattati con soli 30-45 mg al giorno di Metenolone acetato hanno sperimentato una soppressione dei livelli di gonadotropine del 15-65%. (7) Nonostante questa molecola presenti un potenziale soppressivo inferiore se paragonato con altri AAS, la soppressione causata dal Metenolone è comunque presente e verificabile. Se il Metenolone viene utilizzato a dosi moderate per meno di 8 settimane, il recupero ormonale non dovrebbe essere una esperienza prolungata. Per quanto riguarda la forma iniettabile, ad un dosaggio basso di 100-200 mg a settimana (tipico della foto-moda), il Metenolone dovrebbe offrire una soppressione misurabile meno incisiva di una dose eguale di Testosterone e di Nandrolone, probabilmente a causa della sua natura non aromatizzabile.
Per quanto riguarda la forma orale, come per gli altri AAS orali, se ne consiglia l’assunzione lontano dai pasti in quanto studi hanno dimostrato che l’assunzione di uno steroide anabolizzante orale con cibo può diminuirne la sua biodisponibilità.(8) Questo è causato dalla natura liposolubile degli ormoni steroidei, che può permettere ad una parte del farmaco di sciogliersi con i grassi alimentari non digeriti, riducendo l’assorbimento dal tratto gastrointestinale. Per la massima biodisponibilità , questo steroide deve essere assunto a stomaco vuoto.
I dosaggi medi per la forma orale utilizzati in ambito maschile si aggirano tra i 100-200mg/die per 6-8 settimane, mentre per quanto riguarda la dose orale in ambito femminile si aggira tra i 50-75mg/die. I dosaggi medi per la forma iniettabile utilizzati in ambito maschile si aggirano tra i 200mg ed i 600mg a settimana per 6-8 settimane, mentre per quanto riguarda la dose iniettabile in ambito femminile si aggira tra i 50mg ed i 100mg a settimana. Questi dosaggi sono sufficienti per apportare modifiche apprezzabili nel tono e nella massa muscolare.
Il momento della preparazione nel quale generalmente viene utilizzato il Metenolone è nel “Cut”, nel “Pre-contest” o come componente primario di un bridge.
La forma acetata iniettabile (proveniente dal mercato nero) viene ancora utilizzata come “fat burner” locale (iniezioni intradermiche), data la marcata affinità per i recettori androgeni del Metenolone. Infatti i recettori androgeni nel tessuto adiposo stimolano la lipolisi; inoltre questa possibilità di utilizzo, è rafforzata dalla proprietà antiestrogena, comune al Mesterolone e al Drostanolone (entrambi derivati del DHT), derivante dal legame con il coenzima aromatasico P-450. Inoltre, il Metenolone non mostra affinità con i recettori del Progesterone e con i recettori del Cortisolo; il primo è ovviamente un vantaggio (niente attività progestinica) mentre il secondo è uno svantaggio (niente effetto anti catabolico) facilmente risolvibile abbinando la molecola con un altro AAS o con sostanze aventi tale caratteristica. Si afferma che il Metenolone possegga una spiccata affinità con i recettori AR epatici, caratteristica che porterebbe ad un ingrossamento salubre dell’organo (senza fonte certa).
L’emivita del Metenolone Acetato è di circa 4-6 ore mentre il Metenolone Enantato possiede una emivita di circa 10,5 giorni.
Non essendo un AAS particolarmente potente, il suo abbinamento con altre molecole non è di facile scelta. Certamente, non sarebbe correttamente sfruttato se assunto con composti fortemente AR come il Trenbolone, mentre con molecole con attività non-genomica trova il suo migliore abbinamento. In un “Bulk” di un principiante il Metenolone può essere abbinato al Metandrostenolone (Dianabol) mentre in una fase “Cut” può essere abbinato allo Stanozololo (Winstrol). Anche per questo motivo il Metenolone trova la sua migliore applicazione come molecola per “Bridge”. In ogni caso, sarà il vostro preparatore (caldamente consigliato averne uno) a decidere il da farsi secondo il singolo caso.
Gabriel Bellizzi
Riferimenti:
1- https://en.wikipedia.org/wiki/Metenolone
2- Wiechert R. et al. Chem Ber. 93 (1960):1710.
3- Methenolone acetate, Summary of information for clinical investigators, New Brunswick, NJ.The Squibb Institute for Medical Research, May 30, 1962. Methenolone enanthate, Summary of information for clinical investigators, New Brunswick, NJ. The Squibb Institute for Medical Research, April 15, 1962.
7- Comparative studies about the influence of metenoloneacetate and mesterolone on hypophysis and male gonads. Trenkner R, Senge T, Hienz H et al.Arzneimittelforschung. 1970 20(4):545-7.
8- Anabolic Steroids and Sports Volume II. James E. Wright. Sports Science Consultants, Natick, MA 1982.