Gli AAS (in base oleosa e acquosa) possono essere iniettati sottocute, anche se la loro somministrazione principalmente consigliata è tramite iniezione intramuscolare. Le iniezioni di AAS che utilizzano il tessuto adiposo sottocutaneo come luogo di deposito del farmaco modificano soltanto il tasso di rilascio dello steroide dal sito di iniezione, anche se non è stata dimostrata molta differenza dalle iniezioni intramuscolari. Gli individui che …vogliono assumere un AAS tramite iniezione sottocutanea dovrebbero fare attenzione al quantitativo della soluzione, poiché il tessuto sottocutaneo non può contenere un volume di olio iniettato troppo alto senza creare problemi, arrivando a “tollerare” quantitativi decisamente inferiori rispetto alle iniezioni intramuscolari. Per quanto riguarda le affermazioni circa gli ascessi dovuti a tele metodica di iniezione, c’è da specificare che essi si verificano solo se il sito di iniezione è infetto o se il composto è contaminato.
Come risaputo, quando si parla di iniezioni sottocutanee si sta parlando di un metodo di iniezione che prevede la somministrazione di una soluzione (principalmente) a base acquosa nel tessuto adiposo appena sotto la pelle. Anche le soluzioni a base oleosa possono essere iniettate sottocute, ma non è normalmente raccomandato. Le iniezioni sottocutanee sono utilizzate soprattutto per la somministrazione di Insulina, GH (Ormone della Crescita), HCG (Gonadotropina Corionica Umana) e altri peptidi. Le iniezioni sottocutanee sono utilizzate principalmente per iniettare sostanze a base acquosa e composti che richiedono quantità della soluzione molto piccole (1mL o CC o meno), in quanto i siti di iniezione sottocutanei non possono contenere una quantità elevate di soluzione senza problemi. Gli AAS in base acquosa e oleosa possono quindi essere iniettati sottocute, ma prestando attenzione al volume dell’iniezione, iniettando un quantitativo molto inferiore rispetto alle iniezioni intramuscolari. Gli studi hanno mostrato livelli plasmatici stabili in seguito a somministrazione sottocutanea di AAS con efficienza tale ad una somministrazione intramuscolare. (1) (2)
Gabriel Bellizzi
Riferimenti:
1- Subcutaneous administration of testosterone. A pilot study report. Al-Futaisi AM, Al-Zakwani IS, Almahrezi AM, Morris D. Saudi Med J 2006;27(12):1843-6.
2- STABLE TESTOSTERONE LEVELS ACHIEVED WITH SUBCUTANEOUS TESTOSTERONE INJECTIONS. M.B. Greenspan, C.M. Chang. Division of Urology, Department of Surgery, McMaster University, Hamilton, ON, Canada.
L’Omocisteina è un amminoacido solforato di peso molecolare 135,186 che si forma in seguito alla perdita di un gruppo metilico da parte della Metionina, un aminoacido essenziale ossia che necessita di essere introdotto con la dieta.
È un residuo tossico del metabolismo e, una sua elevata concentrazione nel sangue (iperomocisteinemia) rappresenta un fattore di rischio cardiovascolare. (1)
L’iperomocisteinemia induce lesioni endoteliali e disfunzioni endoteliali, incrementa l’attivazione piastrinica nel sito di lesione microvascolare, portando allo sviluppo di aterosclerosi.
L’aumento dei livelli plasmatici di Hcy crea la condizione chiamata iperomocisteinemia (HHcy), caratterizzata da tre varianti; moderata (16-30 µmol/L), intermedia (31-100 µmol/L) e grave (> 100 µmol/L) HHcy.
L’iperomocisteinemia è un sintomo di alcune patologie, ereditarie e non, e di stili di vita errati (2):
omocistinuria (malattia metabolica dovuta a deficit dell’enzima cistationina-β-sintetasi).
carenza di folati, vitamina B12 o vitamina B6 (3)
tabagismo
eccessivo consumo di caffè e di bevande alcoliche
stile di vita sedentario, ridotta attività fisica
esposizione cronica all’inquinamento atmosferico, specialmente al particolato
La mutazione MTHFR (metilentetraidrofolato-reduttasi) che ostacola il processo di trasformazione e causa un aumento di Omocisteina. Si tratta di una mutazione piuttosto frequente (frequenza allelica intorno allo 0.5 nella popolazione italiana). (4) La mutazione (o, meglio, il polimorfismo) più comune è C677T.
In presenza di varie malattie si può registrare iperomocisteinemia, in particolare in caso di ipotiroidismo, psoriasi, lupus eritematoso sistemico, artrite reumatoide. Anche durante trattamenti farmacologici con metotrexate, carbamazepina, fenitoina ed isoniazide inibitori delle COX-2 è possibile l’aumento dei livelli ematici di Omocisteina.
L’iperomocisteinemia in combinazione con altri fattori di rischio (iperlipidemia, ipertensione, fumo, alimentazione scorretta, stress) promuove l’aterogenesi, l’ateromatosi e l’aterosclerosi, che alla fine portano allo sviluppo di coronaropatia , malattie delle arterie periferiche, ictus o trombosi venosa.
Recenti studi hanno dimostrato che un aumento del 25% (circa 3 µmol/L) dei livelli di Omocisteina è associato ad un rischio superiore al 10% che si verifichino eventi cardiovascolari e un rischio che si verifichi un ictus superiore al 20%.
Quando queste concentrazioni superano i 15 µmol/l, vi è un aumento del 25% della mortalità.
Inoltre, l’Omocisteina è un importante fattore di rischio indipendente per lo sviluppo di malattie quali l’Alzheimer, il Parkinson e la sindrome metabolica.
I valori di normalità dell’Omocisteina sono:
valori < 13 µmol/L (uomini)
valori < 10.1 µmol/L (donne)
In caso di omocistinuria da MTHRF i valori ematici sono più alti dalle 10 alle 50 volte, mentre le altre cause e patologie danno aumenti solitamente più contenuti.
L’uso di AAS ha effetti sul lungo termine nelle concentrazioni plasmatiche di Omocisteina.
In tale contesto, l’aumento dei livelli di Omocisteina dipende da:
il tipo di AAS:
Gli AAS metilati in C-17 sono epatotossici e alterano il metabolismo dell’Hcy nel fegato.
Le molecole soggette ad aromatizzazione (Methyltestosterone, Methandrostenolone ecc) o che posseggono attività estrogenica (vedi Oxymetholone) aumentano la concentrazione di estrogeni (E2) o agiscono come tali, e riducono i livelli di Cobalamina (Vitamina B12) e folati.
Una deficienza di folati e B12 nel plasma ha dimostrato di elevare la concentrazione ematica di Hcy.
2. Durata d’uso:
C’è una relazione lineare significativa tra abuso di AAS a lungo termine e iperomocisteinemia.
Somministrazioni oltre i 6 mesi portano ad un aumento dei livelli plasmatici di Hcy.
3. Il dosaggio di AAS:
Gli studi hanno mostrato iperomocisteinemia acuta nei Bodybuilder che usano dosi sovrafisiologiche di vari tipi di AAS.
4. L’uso simultaneo-staking di diversi AAS:
Il meccanismo dell’iperomocisteinemia durante o dopo l’abuso di AAS è multifattoriale.
Gli AAS sono noti per causare (a diverso grado) una marcata dislipidemia, con una significativa riduzione delle lipoproteine ad alta densità (HDL) e un aumento delle lipoproteine a bassa densità (LDL) cosa che porta ad un aumento del rischio di sviluppare eventi cardiovascolari.
Inoltre, gli AAS influenzano il sistema emostatico, attraverso una maggiore attività fibrinolitica e una contemporanea aggregazione piastrinica.
L’attivazione della cascata coagulativa porta ad un tempo di sanguinamento prolungato (INR e PT risultano notevolmente elevati).
Gli AAS influenzano il sistema ematologico portando ad eritrocitosi, stimolando il midollo osseo e promuovendo la sintesi di eritropoietina nel rene.
L’eritrocitosi è associata alla viscosità sanguinea che porta a problemi coronarici, alle arterie periferiche e alla comparsa di trombosi venosa.
La sintesi di Omocisteina avviene negli eritrociti, in tal modo il loro numero elevato contribuisce alla comparsa di iperomocisteinemia.
Nella maggior parte delle preparazioni si nota che gli atleti che utilizzano AAS prestano particolare attenzione all’aumento dei lipidi ematici, all’epatoprotezione e all’aumento dell’ematocrito rivolgendo una scarsa (o nulla) attenzione all’aumento dell’Omocisteina. Ciò rende parziale il tentativo di far fronte agli effetti collaterali derivanti dall’uso di AAS.
Quindi, come fare fronte all’aumento dell’Omocisteina ed evitare l’insorgere di iperomocisteinemia?
Per far fronte all’iperomocisteinemia, l’utilizzatore di AAS deve integrare con vitamine del gruppo B (soprattutto vitamina B6 e B12), Acido Folico e Betaina (Trimetilglicina).
Ovviamente, l’utilizzatore di AAS dovrebbe evitare il consumo di alcol ed evitare tutti gli altri fattori che danneggiano l’endotelio vascolare come il fumo, l’ipertensione e la dislipidemia: questi ultimi due fattori sono più difficili da controllare durante l’uso di AAS ma sono comunque riducibili attraverso un adeguata supplementazione (vedi, per esempio, Riso Rosso Fermentato).
Va notato inoltre che, anche se dopo l’interruzione dell’uso di AAS i livelli di Omocisteina calano, il danno all’endotelio vascolare è irreversibile.
Gabriel Bellizzi
Riferimenti:
gtoul.com
1- O. Nygård, SE. Vollset; H. Refsum; I. Stensvold; A. Tverdal; JE. Nordrehaug; M. Ueland; G. Kvåle, Total plasma homocysteine and cardiovascular risk profile. The Hordaland Homocysteine Study., in JAMA, vol. 274, nº 19, Nov 1995, pp. 1526-33, PMID7474221.
2- Catalano R., Rosso V., Spaccamiglio A., Omocisteina e iperomocisteinemia, flipper.diff.org. URL consultato il 13 novembre 2008.
Con Asse-Ipotalamo-Ipofisi-Testicoli (HPTA) ci si riferisce alla connessione tra ipotalamo, ghiandola pituitaria e testicoli come se queste singole ghiandole endocrine fossero una singola entità. Poiché queste ghiandole spesso agiscono in concerto, i fisiologi e gli endocrinologi ritengono conveniente e descrittivo parlare di esse come di un unico sistema.
L’asse HPTA svolge una parte critica nello sviluppo e nella regolazione di un certo numero di sistemi del corpo, come i sistemi riproduttivi e immunitari. Le fluttuazioni di questo asse causano variazioni negli ormoni prodotti da ciascuna ghiandola e hanno diversi effetti locali e sistemici nel corpo.
In breve, l’asse HPTA rappresenta un sistema di stimolazione/inibizione degli ormoni prodotti dalle rispettiva strutture:
Ipotalamo: GnRH (ormone di rilascio delle gonadotropine; in inglese Gonadotropin-releasing hormone).
Ipofisi (o ghiandola Pituitaria): dalle cellule beta e gamma rispettivamente l’ormone follicolo-stimolante (FSH) e l’ormone luteinizzante (LH).
Come ben sappiamo, diversi AAS sono derivati sintetici del Testosterone, il principale androgeno nei maschi. Il Testosterone sopprime marcatamente l’HPTA, mentre altri derivati lo fanno in misura maggiore o minore.
Gli AAS sono classificati in tre categorie principali:
Derivati del Testosterone (Boldenone, Fluoxymesterone, Methyltestosterone)
Derivati del DHT (Stanozolol, Methenolone, Oxandrolone) e forme sintetiche del Dihydroxytestosterone (Mesterolone, Drostanolone)
Derivati del 19-nortestosterone (Nandrolone, Trenbolone).
I fattori che contribuiscono alla soppressione dell’HPTA sono:
L’origine del AAS
Il tasso di conversione del AAS ad estrogeno, attraverso l’enzima aromatasi in alcuni tessuti (adiposo, mammario)
Dose e tempo d’uso/abuso del AAS
Attività androgena del AAS
Gli AAS che sopprimono maggiormente l’HPTA sono:
Derivati del Testosterone.
Il Testosterone blocca l’HPTA dal primo giorno di utilizzo.
2. AAS altamente estrogenici.
Gli estrogeni (principalmente E2-beta estradiolo) causano un feedback negativo sull’ipotalamo per la produzione di GnRH, che a sua volta stimola LH che stimola la sintesi di Testosterone nelle cellule Leydig nei testicoli. Pertanto, gli AAS fortemente soggetti all’aromatizzazione o che posseggono una attività estrogenica (Oxymetholone, Methyltestosterone, Testosterone, Methandienone) influenzano marcatamente la funzione dell’HPTA.
Un altro motivo per cui gli estrogeni diminuiscono la libido è il fatto che aumentano la concentrazione delle SHBG, che legandosi al Testosterone ne diminuiscono la frazione più attiva (Testosterone libero).
Gli AAS derivati dal 19-nortestosterone sono tendenti ad esplicare un’alta attività progestinica. La iperprolattinemia è associata all’uso di 19-norsteroidi (soprattutto se co-somministrati con forti aromatizzabili). La Prolattina (o PRL) è un ormone polipeptidico di 199 amminoacidi prodotto dalle cellule lattotrope dell’ipofisi anteriore (Adenoipofisi) che ne costituiscono il 20%. Ciò è controverso con la produzione di Testosterone. Pertanto, i livelli elevati di prolattina nel siero sopprimono l’HPTA. Gli effetti collaterali associati con il Progesterone e, quindi, con gli AAS progestinici sono simili a quelli degli estrogeni, compreso il feedback negativo di inibizione della produzione di Testosterone.
Questi farmaci si legano fortemente con il recettore degli Androgeni (AR). Gli AAS attraversano la barriera ematoencefalica e si legano ai recettori sull’ipotalamo. Ciò comporterà una marcata soppressione dell’HPTA. L’attività androgena si traduce nelle caratteristiche sessuali secondarie (crescita dei peli e della barba, allargamento delle spalle e il rafforzarsi dei muscoli, l’ingrandimento del pene, dei testicoli e della prostata.)
Tuttavia, altre funzioni biologiche degli Androgeni coinvolgono:
L’eritropoiesi (stimolazione della sintesi di Eritropoietina nei Reni e aumento delle unità di formazione della colonia eritroide nel midollo osseo e promozione della loro differenziazione nelle cellule responsive dell’eritropoietina)
Effetti anabolizzanti, cioè l’aumento della massa muscolare e della forza, e aumento delle dimensioni delle fibre muscolari. La somministrazione di Testosterone porta alla ritenzione positiva dell’azoto e all’aumenta della sintesi proteica.
Lipolisi (beta ossidazione nei mitocondri nel tessuto adiposo) con riduzione del grasso addominale, e diminuzione dell’insulino-resistenza e aumento dell’insulino-sensibilità. Secondo il dottor Michael Scally, autore del libro “A question of muscle”, gli AAS (che non aromatizzano) potrebbero diventare la soluzione contro l’obesità.
Impatto sul SNC (Sistema Nervoso Centrale) con effetto neurodegenerativo, induzione dell’apoptosi nel ippocampo e ipertrofia dell’amigdala. L’amigdala con l’ippocampo fa parte del sistema limbico, che determina lo stato emotivo. I sintomi clinici della neurotossicità da AAS includono aggressività (tipico effetto “roid rage-bipolar”), ipomania-mania, depressione, disturbo bipolare, comportamento psicotico (idee sbagliate, deliri, e allucinazioni).
Gli AAS con elevata androgenicità sono solitamente soggetti ad elevata attività dell’enzima 5-alfa-reduttasi; la conversione del AAS in DHT influenzerà prevalentemente la ghiandola prostatica e il cuoio capelluto (alopecia). I derivati del DHT non sono soggetti ad aromatizzazione ne posseggono attività estrogenica (tranne rari casi come l’Oxymetholone). Ciò implica praticamente che non vi è alcuna sintesi di beta estradiolo dal AAS, fattore primario nella soppressione dell’HPTA. Il DHT è l’androgeno più forte presente nel corpo, e ha la capacità di sopprime di per se l’HPTA. Tuttavia, il DHT possiede anche proprietà antiestrogene. Un fattore che dimostra l’effetto antiestrogeno del DHT si verifica quando vengono inseriti inibitori della 5-alfa-reduttasi (finasteride, dustateride) per il trattamento dell’ipertrofia prostatica o della calvizie maschile. Gli uomini sottoposti a terapia con inibitori della 5-alfa-reduttasi notano spesso il conseguente sviluppo di ginecomastia (ma anche depressione e la soppressione della libido, poiché il DHT è altamente androgeno). Il perché è di facile intuizione: – conversione del Testosterone in DHT + Testosterone soggetto all’azione dell’enzima aromatasi + mancata azione antiestrogenica del DHT = maggiori livelli di Estradiolo con conseguenti effetti avversi.
Il tempo e la dose d’uso/abuso del AAS provocheranno alla fine un atrofia testicolare (a diverso grado); Questo è un tipico meccanismo omeostatico, regolato dall’HPTA.
Si tenga sempre presente che, tutti gli AAS se assunti in dosi sufficienti per promuovere l’aumento della massa muscolare causano una soppressione del Testosterone endogeno. Senza l’intervento con sostanze Testosterone-stimolante, e una adeguata PCT, i livelli di Testosterone dovrebbero tornare alla normalità entro 1-4 mesi dalla cessione del farmaco. Si noti che un ipogonadismo ipogonadotropo prolungato può svilupparsi secondariamente all’abuso di steroidi, cosa che richiede un intervento medico.
Uno dei miti più duraturi nella storia degli steroidi anabolizzanti è la convinzione che il Dr. John Ziegler abbia creato il Dianabol. La verità è che Ziegler non ebbe nulla a che fare con l’invenzione del Dianabol.
I veri inventori erano chimici organici europei che lavoravano in un laboratorio svizzero.
Il Dianabol (Methandrostenolone o Metandienone) [17beta-idrossi-17alpha-methylandrosta-1,4-dien-3-one] fu sintetizzato da chimici organici che lavoravano per la CIBA Pharmaceuticals in Svizzera. I ricercatori europei che hanno creato il Dianabol probabilmente non sentirono parlare del Dr. John Ziegler, ne prima ne dopo il suo sviluppo, e certamente non collaborarono con lui.
– Allora, chi creò realmente il Dianabol? –
Ai seguenti illustri chimici che svolsero studi e ricerche sugli ormoni steroidei per la CIBA Pharmaceuticals (Svizzera) vanno tutti i meriti per la sua invenzione: Albert Wettstein, Alfred fame, Charles Meystre, Ludwig Ehmann, Ernst Vischer, Hans Peter Frey e Walter Voser. Facevano tutti parte dell’equipe che per prima ha descritto la sintesi del Methandrostenolone sulla rivista scientifica fondata in svizzera Helvetica Chimica Acta.
Vischer E, Meystre C, Wettstein A. Herstellung weiterer 1-Dehydrosteroide auf mikrobiologischem Wege. Helv Chim Acta 1955;38:1502-6.
Meystre C, Frey H, Voser W, Wettstein A. Gewinnung von 1,4-Bisdehydro-3-oxo-steroiden. HeIv Chim Acta 1956;39:734-42.
Gli inventori della registrazione del brevetto degli Stati Uniti per il Methandrostenolone (US 2.900.398) sono elencati come Wettstein, Fame, Meystre, e Ludwig Ehmann della CIBA. La richiesta di brevetto fa riferimento solo al suddetto studio del 1956 dell’Helvetica Chimica Acta. Questi ricercatori non erano scienziati da poco . Wettstein collaborò con il team di ricerca di Leopold Ružička che vinse il Premio Nobel per la Chimica nel 1939 per il loro notevole lavoro sulla sintesi del Testosterone. La scoperta del Dianabol era solo un altro successo nella sua carriera.
– Perché allora il Dr. John Ziegler è stato erroneamente considerato l’inventore del Dianabol? –
Il racconto romanzato di come in una piccola cittadina americana un medico avesse creato uno steroide anabolizzante sintetico per migliorare le prestazioni di sollevatori olimpici americani contro i loro omologhi russi che utilizzavano testosterone fu certamente creata come un’arma al servizio del nazionalismo U.S.A. durante la Guerra Fredda. La disinformazione contribuì a far accrescere la “mitologia steroidea” narrando in continuazione questa storia nelle riviste di bodybuilding e anche attraverso il giornalismo. Anche se il Dr. John Ziegler non fu davvero l’inventore del Dianabol ciò non sminuisce l’importanza che ebbe nella storia dell’uso degli steroidi negli Stati Uniti. Ziegler contribuì a facilitare l’adozione degli steroidi in generale, e del Dianabol in particolare, da parte degli atleti americani.
Ziegler fu la prima persona a somministrare il Dianabol agli atleti competitivi poco dopo la sua introduzione da parte della CIBA nel 1958. Ebbe accesso al laboratorio CIBA a Summit (New Jersey) nel corso degli anni 50’ e somministrava già ai pesisti il Testosterone Propionato per “scopi di ricerca”. Tuttavia, non vi era nessuna ricerca sulla sintesi del Methandrostenolone presso l’impianto della CIBA nel New Jersey: tutte le ricerche sulla sintesi dell’ormone steroideo erano state riservate alla sede centrale in Svizzera. Quando il Dianabol divenne disponibile, la CIBA (New Jersey) chiese a Ziegler di somministrare il Dianabol ai pesisti olimpici sul finire del 1959. Successivamente prescrisse 10mg di Dianabol al giorno (due dosi da 5mg) ai pesisti John Grimek, Bill March, Tony Garcy e Louis Riecke. Essi furono i primi atleti ad usare il Dianabol.
Anche se steroidi anabolizzanti come il Testosterone Propionato, il Methyltestosterone e il Nilevar erano già stati utilizzati da alcuni culturisti della West Coast, quando la CIBA introdusse il Dianabol nel 1958, e il Dr. Ziegler iniziò a prescriverlo, l’uso degli steroidi andò rapidamente ad integrarsi nel bodybuilding e nel sollevamento pesi prima della graduale diffusione in tutti gli sport competitivi.
Il Dianabol emerse come lo steroide preferito tra i culturisti e atleti americani. Attualmente rimane uno degli steroidi più popolari utilizzati dai culturisti contemporanei. Per i milioni di atleti che hanno utilizzato il Dianabol, John Ziegler continuerà certamente a detenere un posto speciale nei loro cuori.
Come risaputo, il fattore primario che rende un AAS epatotossico è la presenza di una metilazione in C-17. L’esposizione prolungata nel tempo o ad alte dosi a molecole con tale metilazione può causare danni al fegato.
Per analizzare e trovare una soluzione al problema, bisogna necessariamente partire dalle basi.
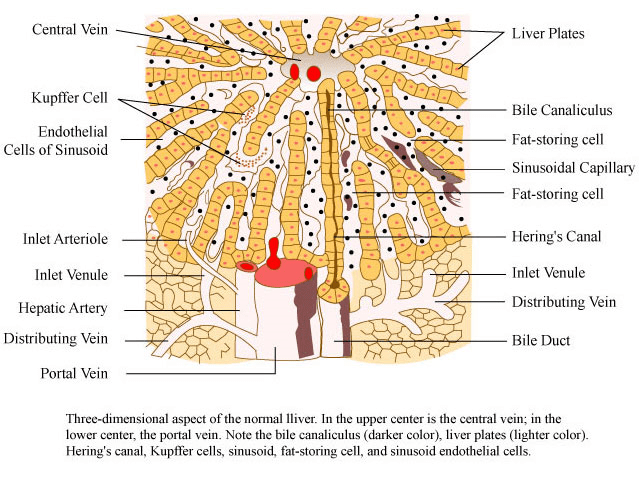
Il parenchima epatico è la parte del fegato che filtra le tossine presenti nel sangue. In sostanza, è un laboratorio biochimico, in cui avvengono diversi processi:
1.Produzione della bile, responsabile per la digestione degli acidi grassi 2. Biosintesi dei fattori di coagulazione (vitamina K), responsabile dei meccanismi di coagulazione 3.Sintesi della maggior parte delle proteine che circolano nel plasma, tra cui l’albumina e la maggior parte delle globuline. 4.Deposito di glicogeno come approvvigionamento energetico (glicogenesi), scomposizione del glicogeno in glucosio (glicogenolisi) e sintesi di glucosio da fonti non glucidiche (gluconeogenesi) 5.Sintesi delle lipoproteine (HLD & LDL), responsabile per l’eterogenesi -amartomatosi infiammatoria nel endotelio arterioso. 6. Produzione ormonale ( IGF1 – Somatomedina C) 7.Deposito di Vitamine (Cianocobalammina-B12) 8.Pulizia e disintossicazione da sostanze chimiche (paracetamolo, farmaci antinfiammatori non steroidei-NSAID, glucocorticoidi, alcool-etanolo, AAS).
Aspetto tridimensionale del fegato normale di MIT OCW.
Ogni volta che una sostanza chimica entra nella circolazione passando in seguito per la vena porta del fegato, l’organo deve rilevare se questa è una sostanza tossica o qualcosa di utile per il sistema. Il fegato metabolizza molti farmaci che non causano problemi epatici. Tuttavia, alcuni AAS possono essere responsabile di una vasta gamma di tipi di problemi epatici e lesioni all’organo. I marcatori biochimici come gli enzimi epatici, noti come transaminasi (alanina transaminasi -ALT / SGOT e aspartato transaminasi-AST / SGPT), fosfatasi alcalina (ALP), γ-glutamil transpeptidasi (GGT), lattato deidrogenasi (LDH) e bilirubina ) sono spesso usati per indicare il danno epatico. Il danno epatico può essere definito da un aumento del:
• (a) livello della ALT superiore di tre volte il limite superiore al normale,
• (b) livello della ALP più del doppio del limite superiore del normale, o
• (c) livello totale di bilirubina superiore al doppio del limite superiore al normale, quando è associato ad un aumento ALT o ALP. Il danno epatico è ulteriormente caratterizzato nel tipo epatocellulari (elevazione ALT) e nel tipo colestatico (aumento ALP).
Gli AAS secondo la loro tossicità epatica, primariamente, sono classificati in due categorie principali:
• (a) Gli AAS metilati in C-17 (la maggior parte dei composti orali),
• (b) Gli AAS non metilati in C-17 (la maggior parte dei composti iniettabili).
Uno dei rari AAS iniettabili metilati in C-17 è lo Stanozololo (Winstrol Depot), mentre i maggiori composti orali non metilati in C-17 sono:
Mesterolone (Proviron)
Methenolone (Primobolan Acetato)
Testosterone Undecaonate (Andriol)
Tra gli AAS metilati in C-17 più forti troviamo:
Methyltrienolone (M3)
Fluoxymesterone (Halotestin)
Oxymetholone (Anadrol 50)
Methandrostenolone-Methandienone (Dianabol)
Methyltestosterone (Teston)
Stanozolol (Winstrol)
Methyldrostanolone (Superdrol)
Methylstenbolone (Ultradrol)
Methy-1-testosterone (M1T)
Methylhydroxynandrolone (MOHN)
Methylepitiostanolo (Epistane)
Chlorodehydromethyltestosterone (Oral Turinabol)
Mibolerone (Cheque Drops)
Tetrahydrogestrinone (THG)
Un caso a parte nei composti metilati in C-17 lo fa l’Oxandrolone, per via del suo assorbimento non a totale carico del fegato.
Tutti gli AAS sono responsabili della distorsione dell’indice ateromatico e dell’alterazione del rapporto delle lipoproteine (HDL / LDL). Questo eventualmente può portare allo sviluppo di malattie cardiovascolari (CVD). La metilazione della molecola steroidea assicura ad essa la capacità di resistere a qualsiasi ambiente ostile, come i succhi gastrici (acido). Quando la sostanza entra nel parenchima epatico, attraverso la circolazione, il fegato deve metabolizzarla. La metilazione è qualcosa di difficile da scomporre, pertanto questo mette sotto stress gli epatociti. Ciò può comportare un aumento dei livelli sierici di AST e ALT (> 100).
Gli AAS iniettabile che presentano una metilazione in C-17 (come lo Stanozololo iniettabile) mostrano una tossicità epatica leggermente inferiore. Una delle ragioni per cui ciò avviene è dovuta al fatto che la molecola così somministrata bypassa il passaggio attraverso la vena porta del fegato (e la conseguente disattivazione di primo passaggio) entrando nel flusso ematico dalla circolazione intramuscolare (via parenterale). Ciò influenza indirettamente il fegato, quindi lo stress è ridotto. Oltre allo Stanozololo, vi sono altri AAS iniettabili metilati in C-17 e venduti nel mercato nero (vedi, ad esempio, l’Oxymetholone e il Methandrostenolone iniettabile). Un metodo complicato affinché un AAS orale metilato risulti meno tossico per il fegato implica che il farmaco venga assunto per via sublinguale, cioè sotto la lingua. Questa specifica zona nella cavità orale è piena di piccoli vasi sanguigni. Si verifica di conseguenza uno spiccato assorbimento del farmaco; quindi lo steroide entra direttamente nel flusso sanguigno, in modo simile a quanto avviene con la somministrazione intramuscolare. In questo modo, la sostanza non raggiunge concentrazioni elevate a livello epatico come dopo somministrazione orale causando un minore stress epatico (anche se ancora significativo).
C’è da aggiungere che, molecole prive di metilazione in C-17 possono, a diverso grado, causare stress epatico: per esempio, il Trenbolone, che non presenta alcuna metilazione, possiede comunque un forte livello di resistenza alla disattivazione epatica, e una significativa tossicità epatica è stata osservato nei Bodybuilder che abusano del Trenbolone. Anche il Boldenone Undecilenato ha mostrato, in studi su animali, di causare danno epatico transitorio, nonostante anche questa molecola sia priva di una metilazione in C-17.
Gli AAS sono stati implicati in quattro distinte forme di lesione epatica:
1. Epatite farmacologica; In questo caso, le transaminasi epatiche sono elevate (> 100). Queste elevazioni sono attribuite all’assunzione di AAS orali, e sono di solito asintomatiche, transitorie e i livelli ritornano alle concentrazioni di base entro alcune settimane dalla cessazione del farmaco. Tali elevazioni sono state maggiormente legate ad AAS quali Fluoxymesterone e Oxymetholone.
Occasionalmente questo aumento è erroneamente diagnosticato come danno muscolare / rabdomiolisi (CPK> 500), piuttosto che come disfunzione epatica, in quanto sia gli epatociti che le cellule muscolari possiedono recettori per gli enzimi SGOT e SGPT. Recentemente, gli studi hanno dimostrato che il GGT è l’enzima più distintivo per la rilevazione della disfunzione epatica.
2.Colestasi; una condizione in cui si verifica ittero. In questo caso non vi è una corretta eliminazione della bile, attraverso il canale biliare e i tubuli biliari intracellulari del parenchima epatico. L’insorgenza è di solito accompagnata da sviluppo di nausea, stanchezza e prurito seguite da urine scure-marroni (urobilinogeno elevato) e ittero (bilirubina elevata). L’ittero può protrarsi anche dopo l’interruzione dell’assunzione di AAS. Tipicamente, sono presenti elevazioni seriche di marcatori colestatici come ALP, γGT, e la bilirubina diretta / indiretta. La biopsia del fegato mostra tipicamente la colestasi con infiammazione, necrosi epatocellulare e iperplasia degli epatociti. Questo fenotipo clinico di colestasi è tipico dell’uso/abuso di AAS.
3.Peliosi epatica; è una rara sindrome in cui i lobi del fegato sono coperti da noduli che contengono cisti piene di sangue. Il fegato può essere ingrossato, e di colore rosso intenso. I livelli degli enzima nel siero sono solitamente normali o lievemente elevati. I pazienti possono lamentare dolore al lato superiore destro. Questa è una condizione critica e talvolta fatale. Tuttavia, la peliosi epatica associata ad abuso di AAS ,in genere, subisce un inversione quando il paziente interrompe l’uso/abuso di AAS.
4.Carcinoma epatocellulare; la complicazione più grave dell’utilizzo di AAS è lo sviluppo di tumori epatici, adenoma (HCA) o carcinoma epatocellulare (HCC). E’ generalmente diagnosticato tramite esame obiettivo, esame del sangue, TC (tomografia computerizzata), ecografia, risonanza magnetica, angiografia epatica e biopsia. Anche se nelle fasi iniziali non dà alcun segno di sé, via via che la malattia si diffonde, però, iniziano a comparire i sintomi specifici, tra i quali il dolore alla parte superiore dell’addome, che si può irradiare anche alla schiena e alle spalle, l’ingrossamento del ventre, la perdita di peso e di appetito, la nausea, il vomito, la sensazione di sazietà, la stanchezza, l’ittero (ovvero il colore giallo della pelle), la colorazione scura delle urine e la febbre. Mentre il Fluoxymesterone è associato alla formazione di HCA, altre sostanze come l’Oxymetholone e il Methyltestosterone possono portare allo sviluppo di HCC. Ci sono diverse strategie terapeutiche per il HCC senza metastasi. In generale il trapianto di fegato è la terapia di scelta per i pazienti selezionati con HCC senza la possibilità di metastasi extraepatica. L’abuso di AAS per un lungo periodo di tempo è un fattore di rischio per lo sviluppo di HCC e quindi gli utilizzatori devono essere ben monitorati. L’ecografia epatica periodica sembra essere una procedura di screening adeguata per rilevare lo sviluppo di lesioni epatiche.
La steatosi epatica è una condizione dovuta principalmente a motivi metabolici. L’obesità e la sindrome metabolica di solito portano all’accumulo di grasso nel parenchima epatico. In alcuni casi, il fegato grasso può essere accompagnato da infiammazione epatica e morte delle cellule epatiche (steatoepatite). Questo caso è reversibile, con un’adeguata alimentazione e l’integrazione di fattori lipotropici (colina, inositolo). La supplementazione fornisce una prevenzione medica e assicura che gli enzimi epatici non siano molto elevati. L’acido Ursodeossicolico (UDCA) sembra essere estremamente utile nei casi di colestasi, in cui avviene l’ittero. Questi sali biliari hanno la capacità di ridurre la concentrazione di bilirubina, diminuire la tossicità del pool biliare e i valori colestatici inversi (ALP, GGT).
Altri potenti antiossidanti, come il Glutatione (iniettabili) o la N-Acetil-Cisteina (NAC), sono inoltre utili. La NAC è un precursore del Glutatione e serve come agente disintossicante del fegato. La NAC serve ad aumentare le riserve di Glutatione nel corpo, insieme con il Glutatione stesso; entrambi si legano direttamente ai metaboliti tossici. Prodotti erboristici come la Silimarina (Cardo Mariano) e il carciofo sono noti come potenti antiossidanti in particolare a livello epatico. Anche l’Acido Alfa Lipoico è un potente agente contro i radicali liberi e lo stress ossidativo. Il Tarassaco è un’erba diuretica che ripulisce dalle tossine attraverso l’urina. Ci sono diversi prodotti contenenti formule per la protezione epatica sul mercato. Personalmente, preferisco evitare i preparati commerciali a causa della loro inadeguatezza nei dosaggi dei vari componenti (cosa spesso riscontrata). Prediligo la creazione di formule per l’epatoprotezione basate sulle esigenze del caso (dal tipo di ciclo e dalle molecole utilizzate). Un’ottima formulazione che ha dimostrato un enorme potenziale prevede l’uso di Silimarina (400-600mg), NAC (3g) e Deursil (300-450mg) a dosaggi adeguati. Tuttavia, queste formulazioni devono essere utilizzate con una dieta bassa in grassi saturi, senza il consumo di alcool né un consumo eccessivo di acetaminofene / paracetamolo. Almeno mentre il soggetto in questione è sotto ciclo di AAS.
L’influenza dell’enzima aromatasi nel mantenimento della salute ossea androgeno-indotta dopo la maturità scheletrica rimane poco chiara. Lo scopo dello studio qui esposto era quello di determinare se l’attività dell’aromatasi è essenziale per il mantenimento delle ossa androgeno-indotto. Nello studio (te…st esatto di Fisher) 344 ratti maschi di dieci mesi di età (n = 73) sono stati assegnati in modo casuale a ricevere un finto intervento chirurgico, una orchiectomia (ORX), ORX + anastrozolo (AN; inibitore dell’aromatasi), ORX + testosterone enantato (TE, 7.0 mg / sett) , ORX + TE + AN, ORX + trenbolone enantato (TREN, non aromatizable, analogo del testosterone non estrogenico; 1,0 mg / settimana), o ORX + TREN + AN. Gli animali esposti a ORX hanno mostrato indici istomorfometrici di elevato turnover della osteopenia e una riduzione del volume osseo spongioso rispetto al gruppo sottoposto all’intervento chirurgico falso. In entrambi i gruppi dove è stato somministrato TE e TREN è stato soppresso il turnover osseo spongioso impedendo totalmente la perdita di tessuto osseo spongioso ORX-indotto. Gli animali ai quali è stato somministrato TE e TREN hanno mostrato anche una maggiore resistenza al taglio del collo del femore rispetto agli animali sottoposti a ORX. Nel gruppo soggetto alla co-somministrazione di AN, gli animali hanno mostrato una leggera inibizione della soppressione del riassorbimento osseo quando trattati con TE, ma senza alterazione della soppressione TE-indotta sulla formazione ossea o gli effetti osteogenici di questo androgeno. Negli animali trattati con TREN, la co-somministrazione di AN non ha prodotto effetti distinguibili sul turnover dell’osso spugnoso o sul volume osseo. Animali sottoposti a ORX hanno mostrato anche una riduzione del muscolo elevatore dell’ano/bulbocavernoso (LABC) e della massa muscolare con una elevata adiposità viscerale. Al contrario, la somministrazione di TE e TREN ha prodotto effetti mio tropici potenti nel muscolo LABC mantenendo la massa grassa a livello degli animali sottoposti al finto intervento chirurgico. La co-somministrazione di AN non ha alterato gli effetti androgeno-indotti sui muscoli e il grasso. In conclusione, gli androgeni sono in grado di indurre effetti diretti sul muscolo-scheletrico e sul tessuto adiposo, indipendenti dall’attività dell’aromatasi.
Gabriel Bellizzi
Fonte dello studio:
J Bone Miner Res. 2014 Nov;29(11):2405-13. doi: 10.1002/jbmr.2265.
Influence of aromatase inhibition on the bone-protective effects of testosterone.
La prima cosa da tenere realmente in considerazione nell’uso del GH per la perdita di grasso è, ovviamente, il dosaggio. La tolleranza individuale al GH può variare notevolmente da persona a persona ma, in linea di massima, la maggior parte delle persone possono utilizzare fino a 14 UI totali a settimana senza che si sviluppi neuropatia (intorpidimento, debolezza o dolore da compressione del nervo a causa della crescita della cartilagine.) Alcuni posso tollerare dosi maggiori, altri invece non tollerano nemmeno il dosaggio prima esposto. Per quanto ne so tutti possono tollerare almeno 7 UI a settimana, e quasi tutti possono tollerare 10,5 UI /settimana, ovvero una media di 1,5 UI / die.
Come prevedibile che sia, maggiore è la dose settimanale e maggiore è il beneficio sulla perdita di grasso.
C’è una vasta gamma di teorie sulla tempistica della somministrazione delle dosi di GH a fine lipolitico (e non). Personalmente, non sono convinto in “assoluto” da nessuna di queste teorie, per il semplice fatto che ho visto ottenere buoni risultati da una vasta gamma di metodi diversi tra loro. Se dovessi scegliere una tempistica per quello che concerne l’uso del GH a fini puramente lipolitici, direi che la somministrazione giornaliera è nettamente superiore a quella, per esempio, che consiste nella somministrazione di 4UI ogni due giorni. Tuttavia, la differenza nei risultati della perdita di grasso nei singoli individui, o le differenze nello stesso individuo con tempistiche di iniezione diverse, sono abbastanza grandi e la differenza tra i protocolli sembra così piccola da non spingermi ad insistere sul fatto che l’uso-almeno-quotidiano è migliore per la perdita di grasso .
Personalmente, e non sono l’unico a pensarlo, credo che non vi sia importanza sulla somministrazione di GH per quanto riguarda i pasti. Comunque, la somministrazione mattutina può risultare una scelta migliore rispetto alla somministrazione serale, o nel corso della giornata per via della possibilità di poter causare una minore soppressione della produzione endogena di GH; in ogni caso i risultati si mostrano migliori con questa metodica. Qualora si volesse somministrare il GH dopo un allenamento, anche questo è un metodo ben collaudato e dagli esiti favorevoli.
Qualora si desiderasse essere nelle migliori condizioni per una data particolare, si rivela di solito accettabile aumentare la dose del 50% nelle due settimane prima di tale data. Anche se questo dosaggio può essere al di sopra della dose accettabile sul lungo termine, il periodo di tempo è comunque abbastanza breve per impedire lo sviluppo della neuropatia, e se essa si manifesta si rivelerà reversibile, sempre a causa del breve periodo di tempo di somministrazione.
In conclusione, vorrei ricordare che la ricerca ha mostrato che la somministrazione di 2UI di GH al giorno possono ridurre le riserve di grasso del 35% quando utilizzate insieme ad una dieta con calorie ridotte.
Un protocollo lipolitico che contempli l’uso di GH può essere esemplificato come segue:
Settimana 1-8: GH 2UI/die divise in due dosi (mattino e pre o post-workout)
Settimana 1-2-5-6: Efedrina (20mg x 2 volte al giorno) Caffeina (200mg x 2 volte al giorno)
Settimana 3-4-7-8: Clenbuterolo (40-60mcg/die).*
Anche in questo caso vige sempre la stessa regola: ne poco ne troppo vanno bene, il giusto va bene.
*L’esempio esposto non è in nessun modo un consiglio o un incitamento alla pratica.
Il Meldonium (nome commerciale del Mildronato) è un farmaco dal mercato limitato, sviluppato negli anni 70 da Ivars Kalviņš, e prodotto principalmente da Grindeks in Lettonia e da diversi produttori di farmaci generici. E ‘distribuito nei paesi dell’Europa orientale come farmaco anti-ischemico.[1] Da farmaco di “nicchia” il Meldonium è divenuto dai primi mesi del 2016 il protagonista dei soliti “scandali” sportivi (capri espiatori è un termine più adatto), infatti dal 1 ° gennaio 2016 è stato inserito nella lista WADA (World Anti-Doping Agency) delle sostanze vietate per utilizzo da parte degli atleti.[2] Tuttavia, vi sono dibattiti sul suo uso come sostanza per il miglioramento delle prestazioni atletiche, per valutare oggettivamente questa molecola bisogna innanzitutto conoscerla.
Il farmaco è stato inventato nella metà degli anni 70 del secolo scorso presso l’Istituto di sintesi organica della Lettonia SSR Academy of Sciences da Ivars Kalviņš.
Il nome chimico del Mildronato è 2-(2-Carbossietil)-1,1,1-trimetilidraziniopropionato[3,4], è un analogo strutturale della γ-butirrobetaina, con un gruppo amminico in sostituzione del C-4 metilene della γbutirrobetaina, questa è un precursore della biosintesi della carnitina[5]; in ambito medico, il Meldonium può essere utilizzato per il trattamento delle malattie coronariche.[6,7]
Si pensa che l’azione del Meldonium si esplichi attraverso la sua capacità di aumentare la dimensione dei vasi sanguigni e, quindi, di migliorare il flusso sanguigno.[8] Anche se le osservazioni iniziali hanno suggerito che il Meldonium sia un analogo non competitivo e non idrossilato della γ-butirrobetaina[9], ulteriori studi hanno mostrato che il Meldonium è un substrato per la γbutirrobetaina diossigenasi.[10,11,12]
Analisi con raggi-X cristallografici e studi biochimici in vitro suggeriscono che il Meldonium si lega alla tasca substrato della γ-butirrobetaina idrossilasi e agisce come substrato alternativo e, quindi, si comporta come un inibitore competitivo.[13]
Normalmente, l’azione di questo enzima sui suoi substrati γ-butirrobetaina e 2-chetoglutarato dà, in presenza di un ulteriore substrato di ossigeno, prodotti quali L-carnitina, succinato, e anidride carbonica; in presenza di questo substrato alternativo, la reazione produce acido malonico semialdeide, formaldeide (simile all’azione del istoni dimetilasi), dimetilammina, e (1-methylimidazolidin-4-il) acido acetico, «un prodotto inaspettato con un carbonio aggiuntivo nel legame di carbonio derivante dalla N-demetilazione accoppiata al riassetto ossidativo, probabilmente tramite un meccanismo radicale inusuale.»[14]
Questo meccanismo insolito suggerisce che la molecola possa comportare una reazione di riordinamento di Stevens[13], l’inibizione del meldonium sul γ-idrossilasi butirrobetaina dà un valore medio della massima concentrazione inibente (IC50) di 62 micromolare, che altri autori dello studio hanno descritto come “potente.”[15,16] Il Meldonium è un esempio di inibitore che funge da substrato mimico non-peptidico.[14]
In ulteriori rapporti di ricerca primaria, è stato dimostrato attraverso risonanza magnetica nucleare come il Meldonium abbia la capacità di legarsi anche alla carnitina-acetiltransferasi, un enzima ubiquitario che svolge un ruolo nel metabolismo energetico cellulare; inibisce anche questo enzima, anche se ancora più debolmente (inibizione costante, KI, di 1,6 millimolare).[14][17]
Nel dicembre 2015, in uno studio pubblicato sulla rivista Drug Testing and Analysis, si sosteneva che il Meldonium «dimostra un aumento delle prestazioni di resistenza degli atleti, un migliore recupero dopo l’esercizio fisico, protezione contro lo stress, e un miglioramento delle attività e funzioni del sistema nervoso centrale (SNC)».[18]
Il produttore, Grindeks, ha affermato in un comunicato, che non credeva dovesse essere vietato agli atleti l’uso del Meldonium, e che il farmaco agisce principalmente per ridurre i danni alle cellule che possono essere causati da alcuni sottoprodotti della carnitina. Il Meldonium «è usato per prevenire la morte delle cellule in ischemia e non per aumentare le prestazioni delle cellule normali», dice la nota. «Il Meldonium non può migliorare le prestazioni atletiche, ma può fermare il danno tissutale nel caso di ischemia», che è la mancanza di flusso di sangue ad una zona del corpo.[19]
Come già precedentemente accennato, il farmaco in questione è stato inventato nella metà degli anni 1970 presso l’Istituto di sintesi organica della Lettonia SSR Academy of Sciences da Ivars Kalviņš.[20,21,22] Kalviņš criticò le istanze, dicendo che non si erano presentate prove scientifiche che dimostrassero che il farmaco possa essere utilizzato come sostanza dopante; secondo il “padre” del Meldonium, questa molecola non migliora le prestazioni atletiche in alcun modo, ed era usata dagli atleti per evitare danni al cuore e ai muscoli causati dalla mancanza di ossigeno durante l’esercizio fisico ad alta intensità. A suo avviso, non permettere agli atleti di prendersi cura della propria salute è una violazione dei loro diritti umani, e che la decisione mira a rimuovere gli atleti dell’Europa orientale dalle competizioni e il suo farmaco dal mercato farmaceutico.[23,24]
Liene Kozlovska, il capo del dipartimento anti-doping del centro di medicina dello sport lettone, ha respinto i reclami secondo i quali il divieto è in violazione dei diritti degli atleti, dicendo che Meldonium è pericoloso in dosi elevate, e deve essere utilizzato solo sotto controllo medico per il trattamento di condizioni di salute che ne richiedono l’utilizzo. Ha anche ipotizzato che gli atleti russi potrebbero non aver ricevuto adeguate avvertenze sul fatto che il farmaco sia stato vietato – a causa della sospensione della AntiDoping Agency russa – alla fine del 2015.[25]
Il Forbes ha riportato che il professore di anestesiologia Michael Joyner, presso la Mayo Clinic di Rochester, Minnesota, il quale studia come gli esseri umani rispondono allo stress fisico e mentale durante l’esercizio fisico e le altre attività, ha affermato loro che «le prove sono carenti per molti composti creduti migliorare le prestazioni atletiche. Il suo utilizzo ha una sorta di elemento di leggenda urbana e non c’è molto al di fuori di ciò che dimostri chiaramente la sua efficacia. Sarei scioccato se questa roba [Meldonium] abbia un effetto maggiore della Caffeina o della Creatina (una sostanza naturale che, quando assunta come supplemento, è pensata per migliorare la massa muscolare).»[26]
Ford Vox, un medico statunitense specializzato in medicina riabilitativa ha riferito ad un giornalista che «non c’è molto supporto scientifico per il suo uso come potenziatore della prestazione atletica».[27]
Il Meldonium, che non è approvato dalla FDA negli Stati Uniti, è stato registrato e prescritto in Lettonia, Russia, Ucraina, Georgia, Kazakistan, Azerbaigian, Bielorussia, Uzbekistan, Moldavia e Kirghizistan.[28,19]
Il Meldonium è prodotto da Grindex, una società farmaceutica lettone, con sede in tredici paesi dell’Europa orientale[29] come trattamento per disturbi cardiaci[30,31]; la società identifica il Meldonium come uno dei loro prodotti principali.[32]
Comunque sia, tralasciando i dibattiti tra sostenitori dell’ ”innocenza” o della “colpevolezza” del Meldonium come sostanza dopante, e i discorsi sterili di chi accusa di slealtà gli atleti risultati positivi al farmaco (tra i quali spiccano Maria Sharapova e Alexander Povetkin), questa molecola mostra, secondo il mio personale parere maturato in seguito alla lettura dei dati disponibili, un grande potenziale protettivo più che prestativo, applicabile a diverse discipline sportive compreso il BodyBuilding, il PowerLifting e il Crossfit.
Gabriel Bellizzi
Riferimenti scientifici
1. “Grindeks: We Believe that Meldonium Should not be Included in the List of Banned Substances in Sport“. Grindeks. 9 March 2016. Retrieved 9 March 2016.
2. “Prohibited List“. World Anti-Doping Agency. Retrieved 9 March 2016.
3. Pubchem. “Mildronate“. nih.gov. Retrieved 9 March 2016.
4. Simkhovich et al., “3-(2,2,2-Trimethylhydrazinium)propionate (THP)–a novel gamma-butyrobetaine hydroxylase inhibitor with cardioprotective properties“. Biochemical Pharmacology 1988 Jan 15;37(2):195–202.
6. Sjakste et al., “Mildronate: an antiischemic drug for neurological indications“. CNS drug rev iews 2005 Summer;11(2):151–68.
7. Dambrova et al., “Pharmacological effects of meldonium: Biochemical mechanisms and biomarkers of cardiometabolic activ ity“. Pharmacological research 2016 Feb 2. pii: S1043-6618(15)30171-7
8. “Meldonium“. Retrieved 19 April 2016.
9. Galland et al., “Purification and characterization of the rat liver gamma-butyrobetaine hydroxylase“. Mol. Cell. Biochem. 1998 Jan;178 (1–2):163–8.
11. Henry et al., “γ-Butyrobetaine hydroxylase catalyses a Stevens type rearrangement“. Bioorganic and Medicinal Chemistry Letters 2012 Aug 1;22(15):4975–4978. Epub 2012 Jun 16.
12. Spaniol et al., “Development and characterization of an animal model of carnitine deficiency“. Eur. J. Biochem. 2001 Mar;268(6):1876–87.
13. Leung et al., “Structural and mechanistic studies on γ-butyrobetaine hydroxylase“. Chemistry and Biology 2010 Dec 22;17(12):1316–24.
14. Tars et al., “Crystal structure of human gamma-butyrobetaine hydroxylase“. Biochemical and Biophysical Research Communications 2010 Aug 6;398(4):634–9.
15. For a perspective from a Merck discovery biochemist on the clinical advantages of picomolar to nanomolar (i.e., thousand- to million-fold more potent) inhibitors, see Copeland, Robert A. (2005). “Tight Binding Inhibition; Potential Clinical Advantages of Tight Binding Inhibitors [Chapter 7, §7.8]”. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. Methods of Biochemical Analysis, Vol. 46. New York, NY, USA: John Wiley. pp. 206–209. ISBN 0471723266. [Quoting:] «If one is beginning this pharmacological optimization with compounds displaying very high target affinity, more flexibility in compromising affinity for other parameters can be exercised. Thus, if the starting molecule has picomolar affinity for the target enzyme, and nanomolar affinity will suffice, the researcher can afford to give up 1000-fold in target affinity for the sake of pharmacological optimization. … high affinity of …tight binding inhibitors allows one to minimize the dose of drug to which patients are exposed, thus limiting off-target based toxicities»
16. Rose et al., “Inhibition of 2-Oxoglutarate Dependent Oxygenases“. Chemical Society Rev iews 2011 Aug;40(8):4364-97.
17. Jaudzems et al., “Inhibition of carnitine acetyltransferase by mildronate, a regulator of energy metabolism“. Journal of Enzyme Inhibition and Medicinal Chemistry 2009 Dec;24(6):1269-75.
18. Görgens et al., “Mildronate (Meldonium) in professional sports – monitoring doping control urine samples using hydrophilic interaction liquid chromatography – high resolution/high accuracy mass spectrometry“. Drug Testing and Analysis 2015 Nov-Dec;7(11-12):973-9.
19. “Meldonium Ban Hits Russian Athletes Hard“. Nytimes.com. Retrieved 9 March 2016.
20. “Scientific Board“. osi.lv. Retrieved 9 March 2016.
21. “Ivars Kalv ins: A broad range of medicines based on natural compounds, spearheading a new generation of drugs“. European Inventor Awards. Candidates in the Lifetime Achievement category. European Patent Office. Retrieved 9 March 2016.
22. Niiler, Eric. “The Quirky History of Meldonium“. Wired. Retrieved 9 March 2016.
23. “Изобретатель мельдония назвал две причины решения WADA” [Meldonium inventor named two reasons for WADA decision]. vesti.ru (in Russian). Retrieved 9 March 2016.
24. Kristīna, Hudenko (8 March 2016). “Mildronāta radītājs Ivars Kalv iņš: meldonija pielīdzināšana dopingam ir cilvēktiesību pārkāpums” (in Latv ian). Delfi. Retrieved 10 March 2016.
25. “Antidopinga eksperte: Mildronāts iekļauts aizliegto v ielu sarakstā” (in Latv ian). Diena. 8 March 2016. Retrieved 10 March 2016.
26. Rita Rubin. “Banned drug Sharapova took is widely used, study shows, despite little ev idence that it boosts performance“. Forbes. Retrieved 9 March 2016.
27. Ford Vox. “Sharapova suspension: doping agency’s unfair game of ‘gotcha’?“. CNN. Retrieved 14 March 2016.
28. “Banned Drug Sharapova Took Is Widely Used, Study Shows, Despite Little Ev idence That It Boosts Performance“. Forbes.
29. “Branches and Representative Offices“. Grindeks. Retrieved 9 March 2016.
30. “AS “Grindeks” ir vadošais zāļu ražotājs Baltijas valstīs” (in Latv ian). Grindeks. Retrieved 9 March 2016.
31. Niiler, Eric (9 March 2016). “The Original Users of Sharapova’s Banned Drug? Sov iet Super Soldiers“. Science. Retrieved 9 March 2016.
Per la stragrande maggioranza degli utilizzatori di steroidi anabolizzanti, i tempi di rilevamento degli steroidi non sono di alcuna importanza: dopo tutto, la maggior parte delle persone non sono nemmeno agonisti. Nei soli Stati Uniti, si stima che più di sei milioni di adulti integrino con steroidi androgeni anabolizzanti la loro routine di allenamento per il solo scopo di migliorare le prestazioni, e di questi individui l’85-90% lo fa per motivi personali. Non sono atleti competitivi, essi non svolgono sport a livello agonistico, ma sono semplicemente persone comuni, frequentatori di palestra che stanno cercando di costruire un corpo migliore. Naturalmente, per il restante 10-15%, i tempi di rilevamento degli steroidi possono diventare molto importanti, perché, un test anti doping positivo può comportare la fine della carriera sportiva. Con questa premessa, voglio mostrare i tempi di rilevamento degli steroidi. Ma prima sono necessarie delle informazioni che dovreste sapere.
Il Test Standard
Se si è soggetti a test di rilevamento degli steroidi anabolizzanti, nella maggior parte dei casi, sarà un test che misura il rapporto tra i livelli di Testosterone e di Epitestosterone. In questo caso, se i livelli di Testosterone sono superiori ai livelli di Epitestosterone oltre ad un certo punto, si fallirà la prova e il test risulterà positivo. Nella maggior parte dei circoli sportivi, la linea di riferimento base è 6:1 (Testosterone : Epitestosterone). Tuttavia, ci sono alcune organizzazioni nel mondo che tengono come riferimento un rapporto 4:1.
Al fine di superare tali test, si hanno due opzioni:
nel primo caso ci si deve assicurare che le sostanze assunte siano state eliminate dal corpo in modo tale da garantire i livelli non elevati o alterati durante il test;
nel secondo caso si attua lo stratagemma che per anni, i tedeschi dell’Est, magistralmente, perfezionarono come processo elusivo, somministrando ai loro atleti Epitestosterone; così facendo, essi rimanevano all’ interno del rapporto accettabile di 6:1. I loro livelli di Testosterone erano aumentati, ma al tempo stesso erano aumentati anche i loro livelli di Epitestosterone.
In ogni caso, il test standard è semplicemente un’ analisi delle urine e nulla più. Ci sono test del follicolo del capello che possono essere eseguiti ma, tuttavia, la maggior parte delle organizzazioni sportive non eseguono questi test in quanto sono stati considerati eludibili in molti modi. In ogni caso, quando l’atleta comprende i tempi di rilevamento degli steroidi o di altre sostanze anabolizzanti somministrate, se è a conoscenza di quando si svolgerà il test, che non è difficile da fare, potrà passare il controllo ogni volta senza ripercussioni.
La scelta degli Steroidi
Se si è un atleta agonista, se si ha intenzione di integrare l’allenamento con gli steroidi anabolizzanti o altre sostanze “doping”, è necessario comprenderne a fondo i tempi di rilevamento. Per l’atleta agonista, sarà un po’ limitata la scelta degli steroidi da utilizzare.
Ad esempio, gli steroidi a base di Nandrolone non possono, di norma, mai essere usati da un’atleta agonista dato che il loro tempo di rilevamento si estende oltre un anno. Molti composti di Testosterone devono essere evitati, soprattutto gli esteri o le miscele di Testosterone. Se non siete atleti agonisti allora non importa quello che assumete dato che il problema del test non sussiste.
Il Tempo di rilevamento degli steroidi
Nella tabella qui sotto, troverete i tempi di rilevamento per tutti i più comuni steroidi androgeni anabolizzanti (AAS) e agenti per il miglioramento delle prestazioni ai quali si può avere accesso. All’interno della tabella, ci sono tre categorie; nomi commerciali comuni, la sostanza che è alla base di una tale denominazione commerciale e poi, infine, i tempi reali di rilevazione degli steroidi per l’ormone specifico o sostanza.
Il presente articolo NON costituisce/sostituisce una prescrizione medica, il blog è l’autore dell’articolo declinano ogni responsabilità per quanto riportato nell’articolo, il quale fine è solo quello tecnico-informativo finalizzato a se stesso.
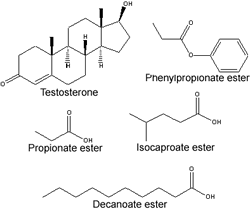
Il Sustanon 250, sia che sia prodotto con questo nome dalla Schering, dalla Organon o da un’altra casa farmaceutica, è uno dei più diffusi e famosi tipi di steroidi anabolizzanti. A differenza di molti altri steroidi iniettabili, il Sustanon comprende una miscela di quattro esteri del Testosterone. In particolare, ogni fiala o ml contiene 30mg di Testo sterone Propionato, 60 mg di Testosterone Fenilpropionato, 60mg Testosterone Isocaproato, e 100mg di Testosterone Decanoato. Questa miscela include esteri a breve, media e a lunga durata d’azione.
Per un ciclo di steroidi, ci sono due vantaggi combinando più esteri nella stessa formulazione come nel Sustanon.
L’utilizzo di più esteri in un’unica soluzione permette un’alta concentrazione totale della molecola di 250 mg/ml senza richiedere una grande percentuale di eccipienti per la solubilità nella soluzione stessa. Questo perché la solubilità dei diversi esteri è pressoché indipendente l’una dall’altra. Così, per esempio, se una soluzione (olio più eccipiente per la solubilità) contiene 100 mg/ml di un singolo estere, potrebbe probabilmente contenerne 200 mg/ml in totale di una combinazione di esteri differenti. La maggiore concentrazione totale aggiunge una maggiore convenienza per l’utilizzatore.
Un secondo effetto di una miscelazione contenente diversi esteri a diversa durata d’azione è che il rilascio degli esteri più veloci porta ad una rapida risposta al prodotto senza attendere la lenta risposta d’azione tipica degli esteri a lunga durata quando usati da soli. Dal punto di vista medico, è auspicabile che il paziente risponda nel più breve tempo possibile al trattamento. Questo discorso vale anche per un ciclo di steroidi. Poiché il Sustanon contiene esteri di breve durata d’azione, può fornire un effetto rapido, fornendo inoltre una piuttosto lunga durata d’azione.
Dal punto di vista del Bodybuilding, questa caratteristica è utile quando un Bodybuilder ricerca uno steroide con una molteplice durata di azione. Inoltre, un prodotto che contiene diversi esteri del Testosterone come Testosterone Enantato o Testosterone Cypionato può facilmente essere utilizzato in un ciclo di steroidi anabolizzanti in abbinamento al Sustanon.
I molteplici esteri presenti nel Sustanon garantiscono un leggero cambiamento dei livelli del farmaco nel tempo. Con un estere iniettato singolarmente, dopo diverse ore o diversi giorni il livello ematico della molecola scende della metà rispetto al totale del prodotto iniettato. Successivamente il livello cala ancora arrivando ad un quarto del livello plasmatico della molecola iniettata in principio; poi il livello scende a un ottavo di quello che era in precedenza, ecc. Questo periodo di tempo è chiamato “periodo di dimezzamento”.
Per il Sustanon non esiste un periodo di tempo determinato per questo. Ritengo che dopo l’iniezione i livelli scendano a metà al 4° giorno, a un quarto al 10° giorno, a un ottavo al 16° giorno, e di un sedicesimo al 23° giorno. Oppure, se si preferisce ragionare avendo come punti di riferimento numeri in termini di percentuale, come valori indicativi i livelli scendono al 40% in 6 giorni, al 30% in 8 giorno, al 20% in 11 giorni e al 10% in 18 giorni.
Come utilizzare queste informazioni in un ciclo di steroidi? Mentre non vi è nulla di certo scritto nero su bianco, una buona scelta sulla quale lavorare è che quando il Clomifene o Tamoxifene è usato correttamente, il recupero della produzione di LH può iniziare quando i livelli di androgeni iniettati sono scesi ad un livello commisurato in 200 mg a settimana. Ovviamente, un recupero più forte si verificherà quando i livelli dello steroide decadono ulteriormente a circa la metà o meno (parlo di una produzione parziale dell’LH).
Quindi supponiamo che il Sustanon è stato somministrato a 500 mg/settimana. In questo caso l’utilizzatore avrebbe bisogno di attendere che i livelli scendano del 40% prima che il recupero possa plausibilmente iniziare. Da quanto riportato sopra, questo avverrebbe all’ incirca al 6° giorno dopo l’ultima iniezione.
Se avessimo come esempio un altro atleta che ha utilizzato un dosaggio piuttosto alto di 2000 mg a settimana, egli avrebbe bisogno di attendere che i livelli scendano al 10% rispetto al prodotto iniettato in principio. Questo avverrebbe all’incirca al 18° giorno dopo l’ultima iniezione del ciclo di steroidi.
Quanto detto riguarda la questione del tempo necessario tra l’ultima iniezione e il punto in cui il recupero incominci ad iniziare.
– La domanda che rimane per quanto riguarda l’insolita vita attiva del Sustanon è, come gestirla? –
Di solito, la determinazione della durata di azione di un farmaco iniettato è abbastanza semplice, essendo calcolata in base all’emivita della molecola e al programma di dosaggio. Tuttavia, il Sustanon non contiene un solo estere, quindi non c’è una risposta matematicamente perfetta. Comunque, possiamo arrivarci abbastanza vicino per scopi pratici.
La quantità settimanale iniettata nel primo giorno dovrebbe essere distanziata dalla successiva, in media, da 5 giorni. Cioè quando la percentuale del farmaco comincia a calare del 40%.
Se per esempio si assumono 750 mg/settimana divisi in tre iniezioni da 250 mg ciascuna, la quantità media giornaliera sarà di 107 mg/die (750 mg divisi per 7 giorni). Quindi la somministrazione ogni cinque giorni sarà modificata affinché il dosaggio settimanale venga assunto entro i sette giorni. Successivamente, le altre iniezioni del ciclo di steroidi, per un livello di mantenimento, saranno tutte da 250 mg.
Questa procedura darà come risultato livelli ematici adeguati molto più rapidamente di quanto non avverrebbe cominciando il protocollo con una singola iniezione settimanale. Per quanto riguarda il dosaggio, ci sono molti modi di vedere le cose, ma uno abbastanza semplice e utile è l’utilizzo del Sustanon partendo e incrementando il dosaggio di 250mg/settimana.
250 mg/settimana
Di solito equivale a niente altro che alla terapia sostitutiva di Testosterone ad alto dosaggio. Non vi è alcuna garanzia che l’uso di tale dosaggio settimanale sarà in grado di portare i livelli di Testosterone sufficientemente al di sopra del range di normalità in tutti i soggetti. Questo dosaggio comunque è abbastanza alto da causare l’effetto collaterale di una produzione di LH inferiore al normale oppure alla sua soppressione, ma nella maggior parte dei casi non è abbastanza alto per fornire effetti anabolizzanti veramente apprezzabili. A seconda della sensibilità individuale, questo dosaggio può essere tale da causare effetti collaterali come ginecomastia se non viene utilizzato un inibitore dell’aromatasi, o può essere sufficiente a causare pelle grassa o acne. Gli effetti anabolizzanti del Sustanon possono essere impressionanti, specie in persone predisposte e con un alto grado di risposta al farmaco. Ma questo non è il risultato usuale per questo livello di dosaggio.
500 mg/settimana
A mio parere, questo è il dosaggio minimo ragionevo le per un ciclo di steroidi. Con questo dosaggio la soppressione dell’ HPTA è più marcata rispetto a dosaggi inferiori, però i guadagni ottenibili sono superiori. Anche in questo caso, dato che il Testosterone aromatizza in estradiolo, per i soggetti sensibili/predisposti l’ utilizzo di un inibitore dell’aromatasi può essere richiesto per evitare effetti collaterali estrogeno-dipendenti. Nessuno, credo, riuscirà a non vedere un sostanziale miglioramento dei guadagni muscolari a questo livello di dosaggio rispetto alla situazione fisica naturale, ma il tasso di miglioramento potrebbe essere lento. Otto settimane, tuttavia, sono sufficienti anche a questo dosaggio per un miglioramento molto significativo, a meno che naturalmente non si è raggiunto il limite di tempo sufficiente nel quale l’utilizzatore con questo dosaggio riesca ad ottenere il massimo.
750 mg/settimana
Vedrei questo dosaggio di utilizzo ideale se si sceglie di fare un ciclo con Sustanon. Se viene utilizzato un inibitore dell’aromatasi è improbabile che si verifichi un aumento degli effetti collaterali rispetto a quelli riscontrabili con un dosaggio di 500 mg/settimana, ed i risultati sono sostanzialmente molto superiori.
1000 mg/settimana
Non ho alcun problema a considerare questo dosaggio molto utile per un sollevatore/Bodybuilder serio che sa con precisione quello che sta facendo. Questo, tuttavia, non è un dosaggio troppo aggressivo per un atleta capace di gestire/prevenire i possibili effetti collaterali. A questo dosaggio, la superiorità sulla formazione di nuovo muscolo è drammatica. Un AI, in questo caso, diventa una priorità per molti utilizzatori.
Infine, ci sono naturalmente le dosi di Sustanon utilizzate di 2000 mg/settimana.
Non vedo un motivo logico per arrivare a tale dosaggio se prima non si è raggiunto il limite di crescita possibile–indipendente da individuo a individuo – raggiungibile con 1000 mg/settimana. In tal caso, se gli obiettivi personali lo richiedono, una dose come questa può essere del tutto appropriata. Indipendentemente dal livello del dosaggio utilizzato, la frequenza delle iniezione deve essere almeno di due volte a settimana, anche se è più preferibile che siano almeno tre le iniezioni settimanali.
Spero ancora una volta di essere stato utile a chiarirvi alcuni dei concetti che stanno alla base della chimica applicata al Bodybuilding.